Chapter 132 Perioperative Management of Severe Traumatic Brain Injury in Adults
Recent estimates of the magnitude of traumatic brain injury (TBI) suggest that each year approximately 1.7 million people sustain a head injury in the United States,1 with about 75% of patients having a concussion or a mild TBI.2 Based on data from the Centers for Disease Control, roughly 52,000 people die annually from TBI, while 275,000 people are hospitalized and nearly 1.4 million people are seen and discharged from an emergency room for TBI. TBI is a contributing factor in 30.5% of all injury-related deaths that occur in the United States, illustrating its importance as a public health problem. TBI is the leading cause of death under age 45 in the Western world.
The impact of brain trauma in the United States and on the country’s healthcare system is highlighted by estimates of direct medical expenditures and indirect costs (e.g., loss of productivity) attributable to TBI being upward of $60 billion in 2000.3 To help perform activities of daily living, 5.3 million Americans depend on long-term assistance, which further underscores the economic and social significance of TBI in today’s society.
Given these demographics, organized efforts to standardize the assessment and management of patients with TBI have resulted in the publication of Guidelines for the Management of Severe Traumatic Brain Injury, which is now in its third iteration.3a A recent cost-benefit analysis showed that widespread adherence and use of these guidelines would result in significant savings: $262 million in annual medical costs, $43 million in annual rehabilitation costs, and $3.84 billion in lifetime societal costs; 3607 lives would be spared annually from mortality due to head injury.4 Ultimately, evidence-based management of patients with severe TBI translates to better patient care, improved outcomes, and a decreased economic and societal burden imposed by TBI.
Initial Neurologic Assessment
Glasgow Coma Scale
The prehospital assessment of a patient’s neurologic condition serves as a guide to the appropriate triage of an individual following a TBI. When transported directly from the scene of injury to a trauma center, patients with severe TBI have lower mortality rates than those who are first taken to nontrauma centers.5 Furthermore, severely brain-injured patients who are admitted and treated at level I trauma centers have improved survival rates and experience better outcomes compared to matched counterparts treated at level II facilities.6
Over the years, many assessment schemes have been created to determine the neurologic status of head-injured patients, with the Glasgow Coma Scale (GCS) being the most commonly used tool to determine the severity of TBI (Table 132-1). Developed in 1974 by Teasdale and Jennett, the GCS gauges a patient’s level of consciousness through an assessment of eye opening, verbal response, and motor response.7 For patients who are mechanically ventilated by an endotracheal tube or via a tracheostomy, thus precluding the patient’s ability to communicate verbally, a GCS score of 1 point is given for the verbal response score with a “T” following to indicate the intubated status of the patient. There are instances in which accurately determining a GCS score is difficult or when the applicability of a score is of limited utility, as with the presence of concomitant facial trauma, ingestion of sedative drugs such as alcohol or barbiturates, or iatrogenic paralysis of patients for intubation. Moreover, physiologic derangements such as hypotension and hypoxia at the time of assessment hinder a patient’s neural response to stimuli; therefore, they represent important factors that may also lead to inaccurate GCS scores, as well as contributing to secondary brain injury.
Table 132-1 Glasgow Coma Scale
| Best Response | Score |
|---|---|
| Eye Opening (E) | |
| Spontaneous | 4 |
| To speech | 3 |
| To pain | 2 |
| Not open | 1 |
| Verbal Response (V) | |
| Conversant | 5 |
| Confused | 4 |
| Nonsense | 3 |
| Sounds | 2 |
| Silence | 1 |
| Intubated | 1T |
| Motor Response (M) | |
| To command | 6 |
| To pain | |
| Localized | 5 |
| Withdrawal | 4 |
| Arm flexion | 3 |
| Arm extension | 2 |
| No response | 1 |
| GCS score = E + V + M (range 3 or 3T to 15) | |
From Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet 2:81-84, 1974.
Severe TBI is classified by total GCS scores of 3 to 8 points, while scores of 9 to 12 points indicate moderate TBI and those between 13 to 15 points denote mild TBI. GCS scores correlate significantly with outcome, with the motor component of the score being most reproducible and yielding the most prognostic information.8 Patients whose GCS scores decrease by 2 points or more between the field and the emergency department are more likely to require surgical intervention.9 Nearly 80% of patients with an initial hospital GCS score of 3 to 5 points have an eventual outcome of death, severe disability, or vegetative state, with a mortality rate of 65% seen in patients with an initial GCS score of 3 points.8,10,11 Among individuals with a GCS score of 3 points on presentation, only 13% have good functional outcomes at the 6-month mark.12 Age, pupil reactivity, radiologic findings on head computed tomography (CT) scan, and extent of extracranial injuries suffered during the trauma serve as additional independent predictors of patient outcome following severe TBI.13–15
Pupillary Examination
The pupillary examination is a critical component of a patient’s initial neurologic assessment following TBI. A dilated pupil that does not constrict in response to light indicates ipsilateral uncal herniation, until proved otherwise. Bilaterally dilated pupils can be seen with hypoxia, hypotension, bilateral oculomotor nerve dysfunction, or severe irreversible brain stem injury. A change in the pupillary examination is the most reliable indicator in determining the side with a mass lesion and carries an 80% positive predictive value. Moreover, pupillary size and reactivity are especially important in the assessment of trauma patients with an admission GCS score of 3 points, because those with bilaterally fixed and dilated pupils have a universally dismal prognosis, with mortality rates of upward of 100%.16–18
TBI Classification
Primary Brain Injury
Primary brain injury refers to damage suffered as a direct result of the initial traumatic event and may be caused by penetrating, blunt, or blast trauma. Bullets and other projectiles often create a significant amount of destruction to brain tissue along their track and can result in injury to brain parenchyma due to fragments of depressed skull. In addition, laceration of cerebral blood vessels can lead to subarachnoid hemorrhage and/or formation of intracerebral hematomas.19 Blunt trauma can result in brain injury because of transmission of force directly at the point of impact or at a point distant from impact due to the displaced brain rebounding against the inner surface of the skull, an event referred to as contrecoup injury.20 The primary injuries of blast TBI are only now becoming understood and represent a regrettable reality of modern warfare and civilian terrorism.
Primary brain injury can be broadly divided into focal and diffuse injuries (Table 132-2). The specific type of injury suffered depends on the nature of the impact during injury (contact or inertial loading), the resultant forces delivered during the impact (rotational, translational, or angular acceleration), and the magnitude and duration of the impact itself.
Table 132-2 Types of Structural Primary Brain Injury
| Focal | Diffuse |
|---|---|
| Hematoma | Concussion |
| Epidural | Multifocal contusion |
| Subdural | Diffuse axonal injury |
| Intracerebral | |
| Contusion | |
| Concussion | |
| Laceration |
Focal injuries include traumatic intracranial hematomas and contusions. Based on data from the Traumatic Coma Data Bank (TCDB), diffuse injuries are more common and constitute upward of 60% of severe TBI, as they span a wide clinical spectrum ranging from concussion to diffuse axonal injury. Higher mortality rates have been seen with focal injuries,21 highlighting the necessity of expedient cranial imaging in evaluating TBI patients via CT scan, which can confirm the presence of a mass lesion and, in turn, prompt emergent surgical intervention if warranted.
Secondary Brain Injury
Secondary brain injury occurs when local phenomena within the skull or systemic factors cause further damage that manifests at the cellular level over the ensuing hours to days following the original insult. The reduction of mortality from severe TBI, even when adjusted for injury severity, age, and other admission prognostic parameters, from 50% to less than 25% over the past 30 years reflects the impact of preventing secondary brain injury specifically through avoiding and treating aggravating factors, such as hypotension, hypoxia, inadequate cerebral perfusion pressure (CPP), and intracranial hypertension.22 These unwanted physiologic perturbations result in cerebral ischemia, which can significantly compound the effects of the initial injury. The decrement in mortality and improvement in outcomes from TBI that has been achieved is largely attributable to the use of evidence-based protocols, which hold avoidance of cerebral ischemia—through intensive monitoring of neurophysiologic parameters and maintenance of adequate cerebral perfusion—as paramount.23,24 Knowing and understanding the pathophysiologic basis underlying cerebral ischemia and intracranial hypertension, the indications for monitoring, and the available treatments directed toward preventing or mitigating their effects are crucial to any neurosurgeon involved in the care of patients with severe TBI.
Cerebral Ischemia
Pathophysiology
At autopsy, 80% of patients who die after TBI have evidence of cerebral ischemia.25 Cerebral ischemia is defined as cerebral blood flow (CBF) that is inadequate to meet the metabolic demands of the brain. While the brain constitutes only 2% to 3% of the body’s weight, it consumes 25% of its oxygen and receives about 20% of its cardiac output. The brain is almost completely reliant on adequate blood flow: nearly 95% of the brain’s metabolism is oxidative without a significant oxygen storage capacity and limited glucose and glycogen reserves. Delivery of oxygen to the brain is contingent on the oxygen content of the blood and CBF; likewise, delivery of glucose and other substrates of metabolism are based on CBF. In turn, a discrete metabolic autoregulatory mechanism exists to couple CBF with the cerebral metabolic rate of oxygen (CMRO2) so that adequate perfusion exists to support local metabolic needs. This principle is illustrated by the Fick equation CMRO2 = CBF × AVDO2, where AVDO2 is the arteriovenous difference in oxygen (in milliliters per deciliter) and is measured by subtracting the jugular venous oxygen content from the systemic arterial oxygen content. Under physiologic conditions, changes in CMRO2 are paralleled by changes in CBF, with AVDO2 remaining constant. In states in which decreased CBF occurs, as in systemic hypotension or when aberrant cerebral pressure autoregulation exists, the brain increases its extraction of oxygen to avoid ischemia. Current evidence suggests that episodes of inadequate CBF, evidenced by low jugular venous oxygen saturation (SjvO2) values of less than 50%, portend bad outcomes, because they are associated with increased morbidity and mortality in patients with severe TBI.26
Two other important homeostatic mechanisms are involved in prevention of cerebral ischemia. The first mechanism indirectly links CBF to CPP via the partial pressure of carbon dioxide (PaCO2) within the arterial system.27 In this process, arteriolar dilation and constriction occurs secondary to changes in PaCO2 and concomitant changes in the pH within the perivascular space, with hypercapnia causing vasodilatation and hypocapnia resulting in vasoconstriction. The second mechanism, referred to as pressure autoregulation, similarly links CBF to CPP.
CBF and CPP
Pressure autoregulation is a fundamental concept in cerebrovascular physiology and is distinct from metabolic autoregulation. Pressure autoregulation allows a constant supply of oxygen and metabolic substrates to be delivered to the brain by maintaining a constant CBF over a range of CPPs. CPP is defined as the mean arterial pressure (MAP) minus the intracranial pressure (ICP). Under normal circumstances, CBF remains relatively constant over a range of CPPs between 50 and 150 mm Hg, ensuring adequate oxygen delivery through a dynamic system of arteriolar vasoconstriction and dilation. Hence, in situations of low perfusion pressures, arteriolar vasodilation occurs, resulting in decreased cerebral vascular resistance and, in turn, increased CBF to normal. On the contrary, when a state of high perfusion pressure exists, arteriolar constriction leads to a decrease in CBF toward normal levels. At CPPs outside the autoregulatory threshold, however, CBF is unable to be maintained within its normal parameters—an exceedingly low perfusion pressure results in arteriolar collapse and a consequent decrement in CBF; hyperemia occurs at high CPPs outside of the limit of autoregulation due to vessels reaching their capacity for vasoconstriction (Fig. 132-1).
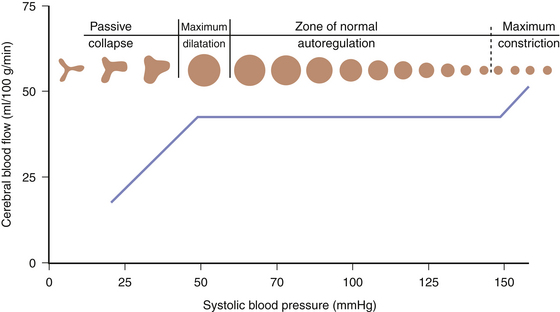
FIGURE 132-1 Cerebral pressure autoregulation.
(From White H, Venkatesh B. Cerebral perfusion pressure in neurotrauma: a review. Anesth Analg 107:979-988, 2008.)
Low CPP has been shown to be associated with higher mortality rates and increased morbidity among survivors following TBI,28–30 especially when systemic hypotension occurs concomitantly.31 Moreover, the normal response of cerebral arterioles to changes in CPP is frequently absent or impaired in 49% to 87% of patients after severe TBI.32–34 Aberrant pressure autoregulation at a range outside normal physiologic parameters may occur after TBI (Fig. 132-2),35,36 with significant consequences. Shifting the lower limit of autoregulation from 50 mm Hg to a supranormal range of 70 to 90 mm Hg could precipitate cerebral ischemia,29,37,38 which is especially noteworthy when considering that up to 60% of patients with severe brain injury experience a reduction in CBF during the first few hours following their trauma.39 Prolonged systemic hypotension or aggressive hyperventilation therapy would further exacerbate ischemia in vulnerable areas and could lead to subsequent cerebral edema. Conversely, autoregulation occurring at a lower-than-typical range of CPPs could result in malignant hyperemia or significant damage to the microcirculation, leading to secondary hemorrhages.40 Thus, absent or impaired cerebral pressure autoregulation represents a particularly significant risk factor for development of secondary brain injury, especially early in the postinjury period.41
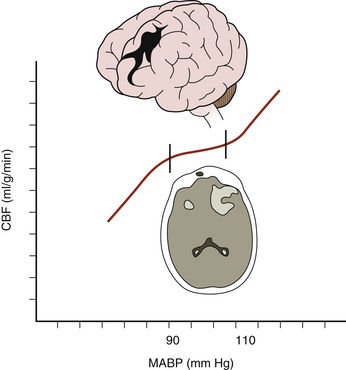
FIGURE 132-2 Aberrant cerebral pressure autoregulation after severe TBI.
(From Rangel-Castilla L, Gasco J, Nauta HJ, et al. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus 25:1-8, 2008.)
Based on the preceding information, CBF is assumed to have a linear relationship with CPP in managing patients with severe TBI. Other key factors that affect CBF include local factors such as physical compression of vessels by mass lesions or edematous brain, reduced cerebral metabolism of oxygen, and post-traumatic vasospasm.42,43 Nearly 33% of severely head-injured patients have CBF values near the ischemic threshold (less than 18 ml/dl) within the first 24 hours after primary injury,44,45 which is seen most often in individuals with diffuse cerebral edema or acute subdural hematomas.46–48 Low CBF values during the first 7 days after injury are associated with significantly higher mortality rates and increased morbidity.32,39,49–51
CPP thus serves as an important gauge of the adequacy of CBF and, in turn, must be monitored and maintained in patients with severe TBI. Through studies of brain tissue oxygen tension (PbtO2) and SjvO2 that correlate with unfavorable outcomes, some investigators have suggested a treatment and maintenance threshold CPP of 60 mm Hg,52,53 while other groups recommend a higher value of 70 mm Hg54,55 to avoid cerebral oxygen desaturation. Based on the results of cerebral microdialysis monitoring, others have discussed a lower CPP threshold of 50 mm Hg.56 In a prospective study in which a CPP-focused management strategy was used, Rosner et al.57 demonstrated a mean mortality rate of 21% in patients with severe TBI when CPP is maintained above 70 mm Hg, as compared to the 40% mortality rate in the TCDB series. Current practice guidelines dictate a threshold around 60 mm Hg due to concern over causing secondary brain injury in patients with aberrant pressure autoregulation, with further adjustments based on ancillary neurophysiologic monitoring of CBF, oxygenation, metabolism and the individual patient’s pressure autoregulatory status.58
Systemic Contributors to Ischemia: Hypotension and Hypoxia
Systemic hypotension and hypoxia have deleterious consequences on patient outcomes following TBI by contributing to secondary brain injury. Episodes of hypotension—defined as systolic blood pressure less than 90 mm Hg; the number of hypotensive events, inclusive of prehospital hypotensive events;13,59 and the increased total duration of such episodes—increase patients’ mortality and morbidity,60 with one study showing a 150% increased risk of mortality in patients with severe TBI who experienced at least one episode of hypotension.61
Similarly, hypoxia portends poor outcomes after TBI. Stochetti et al.62 noted that among severely brain-injured patients, those who had documented oxygen saturations of less than 60% had a mortality rate of 50%, with all survivors being severely disabled, in contrast to a mortality rate of 14.3% and a 4.8% rate of severe disability among nonhypoxic patients. As with hypotension, the number of hypoxic events, along with the duration of hypoxia,60 is predictive of worse outcomes. Establishing an airway early in the postinjury period is one important mechanism to prevent hypoxia; early intubation improves outcomes in patients with severe head injuries.63
Monitoring
Brain Oxygen Monitoring
Brain oxygen monitoring is a means of directly assessing the adequacy of oxygen delivery to brain tissue. Commonly employed modalities toward this goal include SjvO2 sampling via an internal jugular vein catheter directed toward the jugular bulb and direct brain tissue oxygen monitoring by a fiberoptic catheter. Episodes of jugular venous oxygen desaturation (SjvO2 less than 50%) are known to be associated with increased mortality and worse outcomes in patients after severe TBI.26,64 Low values of PbtO2 (less than 10-15 mm Hg) and the extent of their duration (more than 30 min) are also associated with high rates of mortality.65–67 The current guidelines for severe TBI management advocate treatment thresholds for SjvO2 and brain tissue oxygen tension at 50% and 15 mm Hg, respectively.68
Assessment of CBF via Other Modalities
Modalities aimed at directly measuring CBF include placement of thermal diffusion probes and noninvasive techniques such as transcranial Doppler sonography, xenon CT, nitrous oxide uptake, and positron emission tomography. Assessing the metabolic state of the brain via cerebral microdialysis gauges the adequacy of CBF.68 Finally, laser Doppler flowmetry and laser flow flowmetry, performed during surgery with the brain exposed, represent other technologies that measure CBF.69,70
Management Principles
Avoidance of secondary brain injury due to cerebral ischemia is a fundamental principle guiding care following severe head injury. The surgical team must ensure adequate CBF by keeping CPPs around 60 mm Hg, as well as avoiding systemic hypotension, hypoxia, and anemia. Maintenance of CPPs above 70 mm Hg via use of vasopressors and volume expansion has been shown to carry a significantly increased risk of developing acute respiratory distress syndrome, thus obviating the need to aggressively treat patients in such a manner.53,71
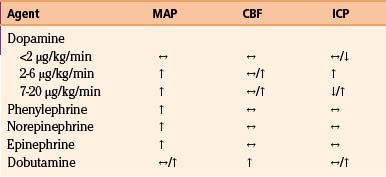
In patients with inadequate CPP, systemic causes of hypotension such as cardiac or spinal cord injury, tension pneumothorax, and bleeding must be ruled out. Intravascular volume must also be assessed via central venous pressure monitoring. Administration of intravenous isotonic fluids is a mainstay of management, but vasopressors are often necessary to maintain adequate CPP. The profiles of commonly used vasopressors are listed in Table 132-3. All listed agents are sympathomimetics, with phenylephrine being the most commonly used.72–75 Though these agents are of significant importance in the management of patients following severe head injury, their use may be associated with complications such as pulmonary edema and/or expansion of preexisting hematomas or contusions76 and therefore must be used with caution.
Delivery of oxygen to brain tissue is predicated on not only sufficient CBF but also oxygen content of the blood. As such, anemia can lead to cerebral ischemia. Despite evidence indicating that low hematocrit is associated with increased mortality77 and morbidity77a in patients with severe TBI, no consensus exists as to a particular threshold for transfusion of blood. While anemia is associated with worse outcomes, there is little support for the aggressive use of packed red blood cells to correct for anemia based on extant data: some studies have noted increasingly negative neurologic outcomes in patients with severe TBI who underwent transfusion,78 while other authors have shown no difference in mortality rates among patients treated under an aggressive transfusion protocol as opposed to those treated along a more restrictive one.79 Interestingly, studies have demonstrated that between 26% and 43% of patients with severe head injuries experience a paradoxical decrement in PbtO2 values in the setting of blood transfusion,80,81 with no effect seen on cerebral metabolism.81
Animal experiments, however, have shown that a hematocrit in the range of 30% to 33% provides an optimal balance between blood viscosity and oxygen-carrying capacity.82 Kee and Wood83 noted that brain tissue oxygen delivery begins to decrease at a hematocrit less than 33%, lending credence to the belief that maintenance of a hematocrit of approximately 30% to 33% is optimal.84 Most centers initiate transfusion of packed red blood cells at a hematocrit of less than 30%. As with all trauma patients, a hematocrit refractory to blood transfusion should prompt a search for extracranial bleeding sources.
Intracranial Hypertension
Pathophysiology
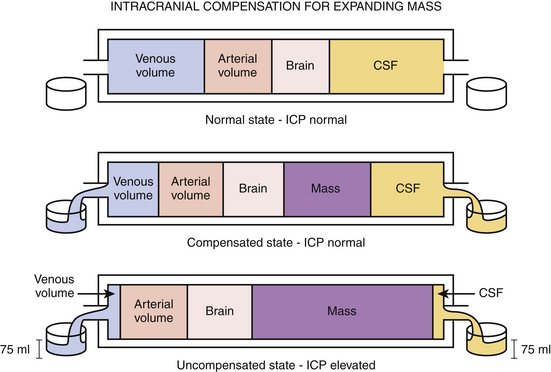
Intracranial contents include blood, brain, and cerebrospinal fluid (CSF). Under physiologic conditions, the intracranial vasculature contains 75 ml of blood, most of which is located within the venous system; CSF occupies a volume of approximately 75 ml and is primarily contained in the ventricles. The intracranial contents are enclosed within the rigid, nonexpansile skull. Based on the principle that the total volume available to intracranial contents is fixed, ICP is determined by the dynamic interplay among the individual components, as delineated by the Monro-Kellie doctrine. Therefore, an increase in an existing component—as seen with hydrocephalus or with post-traumatic cerebral edema—occurs with a concomitant decrease in another. The same concept holds true when a mass lesion occurs, because an additional component has been added to the intracranial space. In this way, in cases of hydrocephalus, post-traumatic cerebral edema, and even new mass lesion development, ICP may remain within normal limits through compensations of the intracranial vascular volume, CSF volume, and brain parenchyma (Fig. 132-3). Physiologic volume-buffering mechanisms exist through which intracranial vascular volume may reduce via increased arteriolar constriction and increased venous outflow, and CSF volume may decrease by being displaced into the thecal sac surrounding the spinal cord. It is estimated that in some instances upward of 100 to 150 ml of new intracranial volume can manifest without any significant increases in ICP, provided that the new mass does not present acutely.85 However, an uncompensated increase in one or more existing compartments or the presence of a new component results in an ICP higher than the normal range of 5 to 15 mm Hg.86,87
CSF normally circulates through the lateral ventricles, the interventricular foramen of Monro, the third ventricle, the cerebral sylvian aqueduct, and the fourth ventricle and out the foramina of Luschka and Magendie. It courses over the spinal cord and the brain and is eventually absorbed into the arachnoid granulations and superior sagittal sinus by means of passive transport. In situations in which there is no intracranial mass lesion, increased ICP following TBI occurs due to perturbations in CSF flow or CBV, as well as because of the development of vasogenic or cytotoxic edema.88 Breakdown products of blood secondary to traumatic subarachnoid hemorrhage preclude the passive absorption of CSF at the arachnoid granulations, which leads to an overall increase in CSF. Increase in CBV occurs when normal pressure and chemical autoregulatory mechanisms are compromised in severe TBI. Vasogenic and cytotoxic edema are the result of a disrupted blood–brain barrier and osmotic dysregulation in ischemic cells.
ICP rises exponentially when an uncompensated state exists. The shape of the pressure–volume curve reflects that as the ICP increases, the brain compliance decreases. The slope of the curve represents the intrinsic compliance of the brain without the influence of ICP. Marmarou et al.89 illustrated this by plotting ICP and volume on a semilogarithmic axis (thus removing the influence of ICP) and obtaining a linear relationship. The slope of the straight line is the pressure–volume index and is the amount of volume necessary to raise pressure 10-fold (Fig. 132-4).
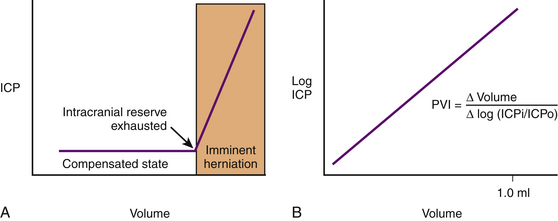
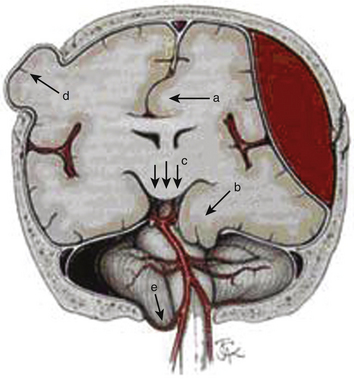
Elevated ICP, known as intracranial hypertension, leads to brain herniation and a decrement in the CPP (since CPP is defined as MAP-ICP). Herniation refers to an event in which brain parenchyma is displaced from its normal location through holes in the dura and skull. This may manifest as subfalcine herniation, uncal herniation, central downward herniation, external herniation of the brain through an open skull fracture, or tonsillar herniation (Fig. 132-5).
Monitoring
Intracranial hypertension is known to have a significant deleterious impact on patient outcome after severe TBI.10,59,90,91 The institution of modern neurocritical care protocols, predicated on monitoring of ICP and other important neurophysiologic parameters, has been credited with improved patient outcomes following severe TBI when compared to historic cohorts.92–94 The question of which patients benefit from ICP monitoring is based on risk stratification for developing intracranial hypertension. The presence of an abnormal head CT is one key factor in predicting which patients with severe brain injuries develop elevated ICPs. Narayan et al.95 found that the incidence of intracranial hypertension in this population is between 53% and 63%. Per findings of the same study and other investigations, individuals with a negative head CT scan on admission following severe TBI have a much lower risk of this occurring, with upper estimates in the range of 10% to 15%.14,96 Interestingly, among this particular subset of patients, the risk of developing intracranial hypertension was comparable to those with abnormal imaging findings if at least two of three risk factors (age over 40 years, unilateral or bilateral motor posturing, or systolic blood pressure less than 90 mm Hg) was present.95
Apart from their basic utility in guiding therapy involving the management of ICP and CPP, ICP monitors may also serve an important diagnostic purpose by indicating the development of worsening intracranial lesions, as a trend of progressively rising ICP values often correlates with the evolution of intracranial pathology.97
ICP Monitoring Devices, Indications, and Techniques of Usage
The Association for the Advancement of Medical Instrumentation has developed the American National Standard for Intracranial Pressure Monitoring Devices, which dictates performance requirements of currently available ICP monitoring devices. These parameters include the ability to (1) measure ICP readings from 0 to 100 mm Hg, (2) be accurate within 2 mm Hg in pressure ranges of 0 to 20 mm Hg, and (3) have a maximum error of 10% in pressure ranges of 20 to 100 mm Hg. Current technology allows for ICP measurement by external strain gauge transducers, microtip catheter strain gauge transducers, fiberoptic transducers, and coupling to a pneumatic transducer. There are many possible intracranial locations in which a particular ICP monitoring device may be placed, including within the ventricle, the brain parenchyma, the subdural space, the subarachnoid space, and epidurally. When considering which device to use, attention must be paid to accuracy, reliability, cost, and potential for patient morbidity. A ventricular catheter coupled to an external strain gauge transducer by fluid coupling is the gold standard.98
In the setting of severe TBI, placement of an external ventricular drain (EVD), or ventriculostomy, for ICP monitoring also allows for CSF diversion and is useful for patients believed to be at risk for development of intracranial hypertension or hydrocephalus. As such, it has a crucial role in guiding therapy, as well as serving as a therapeutic modality by reducing ICP on a sustained basis.99 The most common complications associated with the procedure are hemorrhage, infection, and malfunction. A recent meta-analysis estimated the risk of hemorrhage due to EVD placement as 5.7%, with only 0.61% of hemorrhages being clinically significant100; however, the significantly higher overall hemorrhage rates reported by other investigators highlight that obtaining postprocedural head CT scans may be a confounding factor.101 Lozier et al.’s102 review of ventriculostomy-associated infections (VAIs) cited a rate between 6% and 9%. Other important considerations involving VAIs include the significant association of CSF infections with increasing duration of EVD catheterization, the utility of administering prophylactic antibiotics prior to EVD placement, and the use of antimicrobial-impregnated catheters, which have been shown to reduce the risk of EVD-related infections.103 Based on the previously cited review of VAIs, prophylactic catheter exchange to reduce VAI rates is not efficacious. EVD malfunctions are usually attributable to obstruction in the fluid coupling mechanism by which the external strain gauge catheter transduces ICP, a complication estimated to occur in 2.5% of patients.104
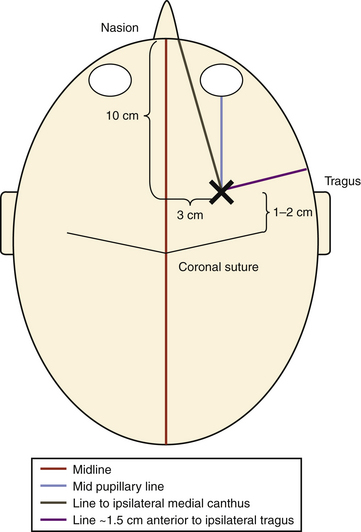
At our institution, right-sided EVDs are preferred to avoid the dominant hemisphere. The patient is maintained in a supine position with the head brought to the edge of the bed; the head of the bed is elevated to roughly 45 degrees. Hair clippers are used to widely shave the head on the same side of the EVD cannulation, and the head is prepared in the standard sterile fashion. Two lines are drawn on the scalp to establish Kocher’s point: one line in the midline from the nasion to a point 10 cm back and another from the previous point to a spot 3 cm lateral to it. This location is where the bur hole placement will occur and should be in the ipsilateral midpupillary line. Finally, two lines are drawn that guide the trajectory of ventricular cannulation: one line from Kocher’s point to the ipsilateral medial canthus of the eye and a second line from Kocher’s point to a point roughly 1.5 cm anterior to the ipsilateral tragus (Fig. 132-6). After administration of a local anesthetic containing epinephrine, a small linear skin incision is made down to bone and the periosteum is scraped off. A twist drill is used to open the cranium, bone fragments and dust are removed, and the dura and pia mater are pierced with a scalpel or sharp object. Using the demarcated lines as a guide, the trajectory at which the drill is used to create the bur hole is the same trajectory used for passing the ventricular catheter: aiming in the coronal plane toward the medial canthus of the ipsilateral eye and in the anteroposterior plane toward a point 1.5 cm anterior to the ipsilateral tragus. This trajectory is exceedingly important because it delineates the trajectory of the ventricular cannulation; if the bur hole is not created in an appropriate trajectory, the EVD cannot be optimally positioned due to its being limited by its bony opening, especially if the cranium is thick (Fig. 132-7). The ventricular catheter is passed to a depth of 6.25 cm using the previously described trajectory. The surgeon may alternatively pass the ventriculostomy using Dandy’s principle, which states that employing a trajectory directly perpendicular to the skull allows ventricular cannulation. Regardless, the ventricular catheter is never passed to a depth greater than 7 cm due to the risk of damaging the brain stem. Once the EVD is in place, it is tunneled through the skin away from the incision through a separate stab incision and is secured in place. A postprocedural head CT is then obtained.
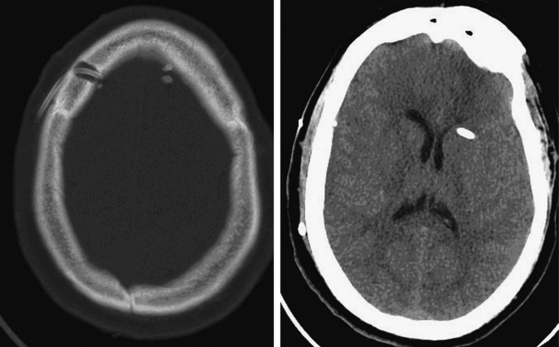
There are cases in which ventricular cannulation can challenge even the most experienced of neurosurgeons. Our experience is to always use the landmarks and trajectory as outlined earlier, regardless of whether a mass lesion or significant shift is present. Most often, errant EVD passes are due to a lateral trajectory (Fig. 132-8), and the neurosurgeon must pay particular attention to avoiding an injury to the ipsilateral internal capsule. In cases in which the ventricle is believed to be cannulated with no egress of CSF, the neurosurgeon may consider attaching a small, plungerless syringe filled with sterile saline to the ventriculostomy catheter. If the ventricle is indeed cannulated, saline should flow from the syringe, through the catheter, and into the ventricle. If the neurosurgeon does not obtain CSF flow through the ventriculostomy after 3 passes, the EVD is left in place and a head CT scan is obtained to determine its position.
Other ICP monitoring devices are frequently used in centers, sometimes as an adjunctive means of monitoring ICP when they are placed in tandem with an EVD and at other times when slit-like ventricles preclude EVD placement. The scalp incision is made in the exact location as for a ventriculostomy, with a smaller drill bit and small stylet used to puncture the dura. When using microtip strain gauge or fiberoptic parenchymal monitors, the probe is advanced between 1 and 2 cm into the brain parenchyma. The microtip strain gauge probe must be tunneled through the skin via a separate stab incision and then be secured in place. However, the fiberoptic monitor can be held in place by a fixation bolt screwed into the skull. The reduced accuracy and reliability, increased propensity for malfunction, and high cost in comparison to EVD placement are factors that must be considered prior to using these alternative ICP monitors.
Management Principles
Current practice guidelines recommend that an ICP treatment threshold of 20 mm Hg be used,105 based on investigations showing that the incidence of morbidity and mortality among severely brain-injured patients is significantly associated with ICPs exceeding 20 mm Hg.59,95,106 The rationale behind treatment of intracranial hypertension is to prevent cerebral ischemia by ensuring adequate CPP and preventing brain herniation. ICP values, however, must be considered as part of the larger clinical picture, together with exam findings and imaging findings, such that established treatment thresholds are considered guidelines that do not preclude treatment at lower values, especially in light of herniation potentially occurring at ICP values less than 20 mm Hg.107
Several methods may be employed to decrease ICP. A stepwise, protocol-based approach to treatment of intracranial hypertension is best suited to prevent the utilization of an excessive number of modalities concomitantly. Maneuvers to optimize venous outflow from the intracranial vascular volume are first used, followed by pharmacotherapy and CSF diversion (Fig. 132-9).
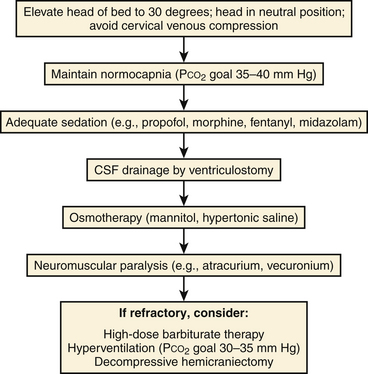
Positioning
Maintaining the head in a neutral position with elevation of the head of the bed to 30 degrees decreases the intracranial blood volume by maximizing venous return from the brain while preserving CPP and CBF.108–110 ICP reduction may thus be readily achieved without increasing the risk of cerebral ischemia.
Cervical Collar Removal
Of pertinence to patients with severe brain injuries is cervical spine clearance, and subsequent cervical collar removal, on ICP reduction. Studies have shown that patients involved in blunt trauma with a GCS score of 3 to 8 points have a higher incidence of cervical spine injuries as compared to other trauma patients,111,112 with rates of 10% to 14%.113,114 Recent investigations have demonstrated the efficacy of a negative helical cervical CT scan in cervical spine clearance among obtunded trauma patients without neurologic deficits that could be potentially explained by spinal cord pathology.115–118 Therefore, cervical spine magnetic resonance imaging (MRI) is unnecessary to remove external cervical orthoses designed for cervical immobilization, which are known to cause a significant and sustained elevation in baseline ICP.119–122 Hence, our institutional policy is that among patients with severe TBIs and no neurologic deficits that can be associated with injury to the spinal cord, cervical collars are expediently removed following a negative helical CT scan of the cervical spine.
Maintenance of Normocapnia
As previously mentioned, alterations in arterial PCO2 result in variations in arteriolar caliber with a consequent effect on CBF and ICP based on the phenomenon of CO2 reactivity. The physiologic mechanism by which the caliber of cerebral arterioles varies involves changes in the pH of perivascular tissue secondary to PaCO2 levels.27 In turn, hypoventilation increases PaCO2 and ICP, whereas hyperventilation reduces ICP at the expense of CBF. Maintenance of normocapnia (PaCO2 between 35 and 40 mm Hg) provides a balance between ensuring adequate CBF to avoid ischemia and preventing the development of intracranial hypertension associated with hypoventilation.
Sedation
A number of pharmacologic agents are used in patients with severe TBI, with the goal of treating pain and agitation and minimizing the effect of a multitude of noxious stimuli, which may contribute to elevations in blood pressure, body temperature, and ICP and resistance to controlled ventilation.123 In addition to using sedative agents in these patients, the surgeon often needs to determine their efficacy and titrate accordingly. Bispectral index monitoring is a useful tool to accomplish this, because it can provide objective information as to the degree of sedation in these critically injured patients.124 The effects of commonly used analgesic and sedative agents on MAP, CBF, and ICP are shown in Table 132-4.
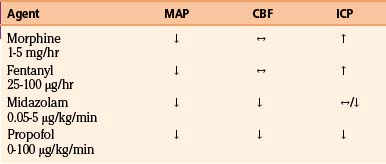
Propofol, a short-acting sedative anesthetic with a rapid onset of activity, is frequently used and has been shown to have superior effects on ICP reduction compared to narcotic-based agents.125 In addition, studies have demonstrated that propofol decreases cerebral metabolism and oxygen consumption, has anti-inflammatory properties, and is an antioxidant, leading to the possibility of its having a neuroprotective effect.126,127
Narcotics have also commonly been employed for providing sedation and analgesia, both alone and with other agents. Morphine sulfate has been shown to provide excellent analgesia but has poor sedative properties. Tachyphylaxis may be seen that can lead to a need for dose escalation and a potential for withdrawal when therapy is discontinued.123 Rapidly metabolized synthetic narcotics such as fentanyl, sufentanyl, and remifentanil are being used more often due to their brief duration of action. However, increases in ICP and decreases in MAP seen with fentanyl and sufentanyl usage mitigate their utility.128,129
Benzodiazepines have the untoward side effect profile of being neurodepressants and exhibiting long half-lives and thus are not considered ideal sedative agents in the brain-injured population. Midazolam, however, is a commonly employed agent in the management of patients with head injuries due to its short half-life compared to other benzodiazepines and its relatively minimal effect on hemodynamics, especially in comparison to propofol. Midazolam exhibits a similar safety and efficacy profile versus propofol,130,131 with a decrement in the cerebral metabolism and oxygen consumption seen with benzodiazepine administration being a potential additional benefit.
Dexmedetomidine, an alpha-2 agonist, is one of the newer agents to be studied and used in neurosurgical patients. Increased CPP and decreased ICP have been seen with its use, which, combined with a benign pharmokinetic profile,132 warrants further study for its use in patients with severe TBI.
Hyperosmolar Therapy
Osmotic Diuretics
Intravenous administration of osmotic diuretics has been used for several decades for reduction of ICP. Originally used by Wise and Chater in the 1950s,133 mannitol not only works to reduce ICP but also results in the improvement of MAP, CPP, and CBF.134 A randomized, controlled trial by Schwartz et al.135 comparing administration of mannitol and pentobarbital in patients with high ICP due to head injury showed superior ICP reduction, CPP preservation, and decreased mortality in those given mannitol. Similarly, improved ICP reduction and better CPP and SjvO2 values were seen in a subset of severe TBI patients who were given intravenous mannitol, in comparison to those treated with an EVD or hyperventilation alone.136
The effect of mannitol on ICP reduction is believed to be attributable to two separate factors, one involving its rheologic effect and the other pertaining to its osmotic effect. The rheologic properties of mannitol account for its ability to rapidly decrease ICP by mediating an expansion in the volume of plasma, which results in a reduction of the hematocrit and increased red blood cell deformability.137,138 The resultant decrease in blood viscosity leads to an increase in CBF and a subsequent decrease in ICP due to arteriolar vasoconstriction, as dictated by pressure autoregulation.139,140 Mannitol is able to reduce ICP over the longer term by creating an osmotic gradient by which water diffuses from the brain parenchyma into the intravascular compartment.141 Current guidelines support the administration of mannitol via bolus dosing of 0.25 to 1 g per kilogram of body weight.142
An important side effect to consider when administering mannitol is the potential for developing acute renal insufficiency secondary to acute tubular necrosis. Serum osmolarity of greater than 320 mOsm is a key risk factor for this to occur.143 Mannitol use may also lead to derangements in a patient’s intravascular volume status and concentrations of various plasma electrolytes; as such, blood urea nitrogen, creatinine, and sodium levels should be monitored closely. In addition, prolonged mannitol administration may result in a reversal of the osmotic gradient due to its crossing the blood–brain barrier and being taken up into edematous brain tissue, a scenario that is more likely if mannitol is administered as a continuous infusion.144
Studies have also shown that the use of furosemide (0.7 mg/kg) as an adjunctive ICP-lowering agent with mannitol elicits a more marked and sustained decrement in ICP.145 The usual intravenous dose is between 20 and 40 mg.
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Tumors Involving the Cavernous Sinus
Tumors Involving the Cavernous Sinus
 Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


