Chapter 203 Peripheral Nerve Tumors of the Extremities
In the United States, it is estimated that annually six out of every 1 million people undergo surgery for peripheral nerve tumors.1 The neurosurgeon is often consulted to diagnose and manage tumors involving the peripheral nerves of the extremities. Intraneural tumors arise from components of the nerve sheath (Schwann cells, perineural cells, fibroblasts), neurons, or invading metastases and cause expansion of the nerve. Extraneural tumors arise from adjacent structures and can compress or infiltrate the nerve. The majority of tumors involving the peripheral nervous system are benign tumors of the nerve sheath (schwannoma or neurofibroma). Tumors can arise sporadically or be associated with genetic syndromes that produce multiple tumors such as neurofibromatosis or schwannomatosis.
Presentation
Patients with a peripheral nerve tumor usually present to clinic with a visible or palpable mass (may be symptomatic or asymptomatic), sensory alterations, pain, weakness, or autonomic dysfunction, or the tumor may be an incidental finding (Table 203-1). Many peripheral nerve tumors are first noticed as asymptomatic lumps. There may be swelling or a history of swelling around the lump. Sensory alterations might consist of anesthesia, hypoesthesia, hyperesthesia, paresthesias, or dysesthesia in the distribution of the affected nerve. Stimulation of the skin with innocuous mechanical or thermal stimuli might elicit pain (allodynia) or patients might have a heightened response to painful stimuli (hyperalgesia). The area surrounding the tumor might spontaneously ache or be tender to palpation. Palpation of the mass might elicit paresthesias or dysesthias. Involvement by the tumor of a motor or mixed nerve might produce frank weakness or, more commonly, subtle functional impairment with fine motor tasks. Autonomic dysfunction can manifest as alterations in the coloration, texture, or temperature of the skin or in the amount of sweat. On occasion, peripheral nerve tumors cause complex regional pain syndrome.
TABLE 203-1 Common Presenting Symptoms of an Extremity Peripheral Nerve Tumor
| Visible or palpable mass |
| Pain |
| Altered sensation (anesthesia, hypoesthesia, hyperesthesia, paresthesia, or dysesthesia) |
| Weakness |
| Autonomic dysfunction (sweating, discoloration, edema, temperature changes) |
| Incidental finding on imaging studies |
History and Examination
A thorough physical examination should be performed on all patients with peripheral nerve tumors. Patients should be examined for diagnostic signs of NF1 (Table 203-2). The neurologic examination includes testing the cranial nerves as well as the motor and sensory function and deep tendon reflexes in all four extremities. A systematic and reproducible examination permits detection of subtle clinical deficits due to the tumor, pathology at higher levels of the neuraxis, or previously undetected tumors elsewhere in the body. Changes in motor function are a late finding; often examination detects only subtle sensory changes or no deficits at all. Discoloration or atrophy of the skin, abnormal sweating, temperature changes, nailbed atrophy, or distal swelling may be findings of autonomic dysfunction.
TABLE 203-2 National Institutes of Health Criteria for Neurofibromatosis Type 155
Imaging
Historically, the diagnosis of peripheral nerve tumors was based on history, physical examination, and electrophysiologic studies. Although soft tissue masses are not visible on plain film radiographs, these images can show secondary skeletal changes due to tumor mass effect or calcifications intrinsic to the tumor. Computed tomography (CT) also can demonstrate secondary skeletal changes and intrinsic calcifications and offers improved resolution of soft tissues. CT is the imaging modality of choice for patients with contraindications to MRI. Since the 1980s, MRI has emerged as the gold standard for imaging peripheral nerve tumors.2 MRI permits detailed visualization of soft tissue at high resolution and can demonstrate the presence of a mass, the anatomy of involved peripheral nerves, and their relationships to adjacent structures such as major blood vessels and joints. The advent of high-resolution MR neurography (MRN) combined with the expansion of the number of neuroradiologists experienced with performing and interpreting these studies has significantly affected the field since the turn of the 21st century.3–5 MRN offers improved contrast and resolution that facilitates determining the precise location, size, shape, and resectability of peripheral nerve masses.5
A combination of T1, T2, STIR (short tau inversion recovery), and gadolinium-enhanced scans provide clues regarding the nature of the tumor and can help distinguish benign and malignant lesions. MRI provides crucial information for preoperative planning and aids in determining the specific pathology of the tumor. Further information can be obtained regarding cellular composition and areas of hemorrhage or cysts. For example, benign nerve sheath tumors are often isointense or slightly hyperintense on T1, hyperintense on T2, and heterogeneously enhancing (Figs. 203-1 and 203-2). The majority of neurofibromas, and some schwannomas, display increased central and decreased peripheral T1 signal intensity (the “target sign”) and an opposite pattern of increased peripheral and decreased central T2 signal intensity.6 Patients with NF1 or schwannomatosis often have multiple tumors throughout their bodies and can have multiple discrete nodules on an affected nerve or develop large plexiform tumors (Fig. 203-3).
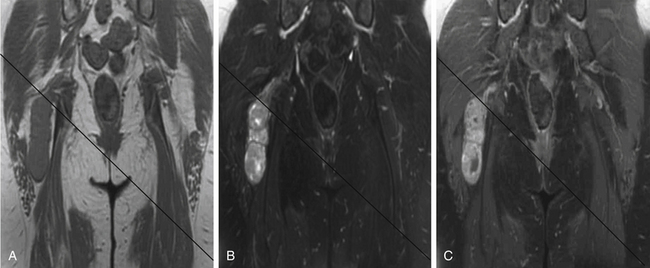
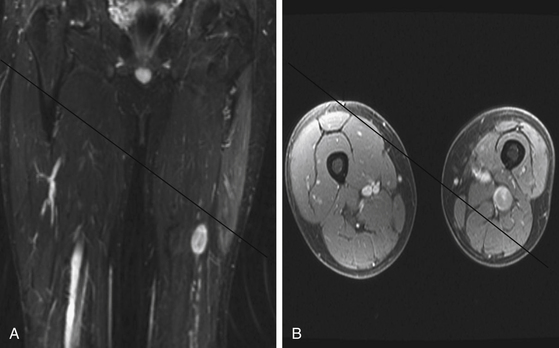
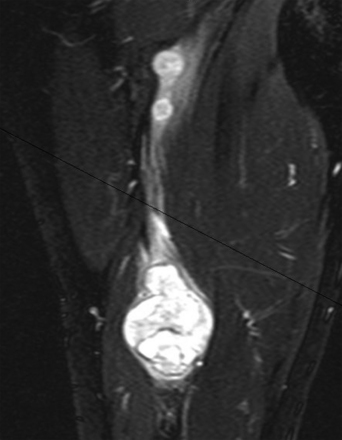
The capacity to accurately assess tumor grade or malignancy based on MRI remains limited. In general, malignant tumors heterogeneously enhance, are less discrete, and are more likely to have areas of necrosis or hemorrhage (Fig. 203-4) However, benign tumors can also have all of these characteristics. A rapid increase in size of a tumor on serial MRI can suggest malignancy, but small increases in tumor volume are difficult to detect.
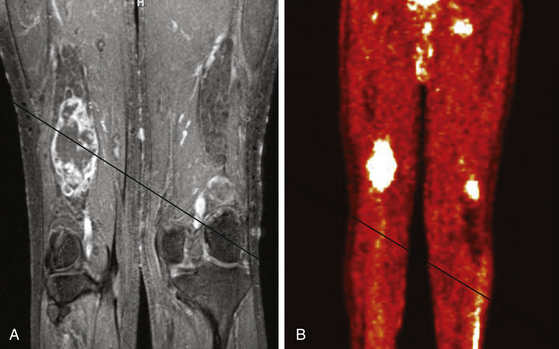
FIGURE 203-4 Magnetic resonance image (MRI) and 18FDG-PET (18fluorodeoxyglucose positron emission tomography) of a malignant peripheral nerve sheath tumor (MPNST) of the sciatic nerve. A, Coronal T1-weighted post-gadolinium image demonstrates a large, infiltrative, heterogeneously enhancing tumor of the right sciatic nerve with areas of necrosis. B, Coronal FDG-PET scan demonstrating marked uptake within the right sciatic nerve tumor.
Metabolic imaging using with 18fluorodeoxyglucose (18FDG) positron emission tomography (PET) often adds valuable information regarding the grade of a tumor (see Fig. 203-4). Ferner and colleagues introduced the use of 18FDG-PET to detect malignant transformation of benign plexiform neurofibromas to MPNSTs.7 Several small series have reported sensitivity ranging from 89% to 100% and specificity ranging from 83% to 95% depending on the time of scanning after injection and the cutoffs used for standard uptake values.8–10 The addition of CT anatomic imaging to 18FGD PET can facilitate targeting biopsies to metabolic hot spots, to further augment diagnostic sensitivity.11
Benign Peripheral Nerve Tumors
The majority of peripheral nerve tumors are benign. Intraneural tumors can arise from the nerve sheath (schwannoma, neurofibroma, intraneural perineuroma) or other intrinsic structures of the nerve (desmoid tumor, intraneural ganglia, lipomatous tumors, ganglioneuroma, metastases, and others). Extraneural tumors represent a diverse population of entities that can arise from or metastasize to adjacent soft tissue, bones, or joints and compress or invade peripheral nerves. This section focuses on schwannomas and neurofibromas.
Schwannoma
Schwannomas (neurilemmomas) are benign tumors that arise from the nerve sheath. They are typically solitary and slow growing. A schwannoma can arise from any peripheral nerve with a Schwann cell, including distal portions of cranial nerves. Sporadic schwannomas can occur at any age but are most common in the third to sixth decades of life.12
Bilateral acoustic schwannomas are pathognomonic for the diagnosis of NF2, but there is no association between peripheral schwannoma and NF1.13 Since the turn of the 21st century the diagnosis of schwannomatosis has gained acceptance for patients with multiple peripheral nerve schwannomas in the absence of acoustic schwannomas. The specific diagnostic criteria for schwannomatosis are listed in Table 203-3. The annual incidence of new diagnoses has been estimated to be 1 in 1,808,300 in a Finnish population-based study, which would correlate to a birthrate of about 1 in 80,000.14 Schwannomatosis usually occurs sporadically, but cases of autosomal dominant transmission have been reported.15 The tumors that develop may be conventional globular schwannomas or plexiform and arise throughout the body or may be limited to a single segment or nerve. It is currently unknown whether the diagnosis of schwannomatosis alters the risk of malignant transformation of a schwannoma.
TABLE 203-3 Diagnostic Criteria for Schwannomatosis56
Pathology
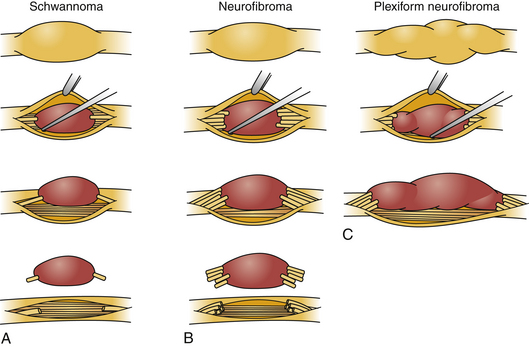
Schwannomas can be broadly classified into four subcategories: conventional schwannoma (the focus of this section); cellular schwannoma, a benign tumor that histologically resembles MPNST; plexiform schwannoma, which simulates plexiform neurofibroma or in its cellular form, MPNST; melanocytic schwannoma, which is often mistaken for a melanoma.16 Conventional schwannomas are usually globoid and homogeneously firm, but they can have ballotable cystic areas. On gross examination the tumor has a well-defined capsule, appears shiny, and is tan-yellow. Histologically, they are distinguished from other nerve sheath tumors by being composed exclusively of Schwann cells; perineural cells and fibroblasts are not present.17 Diagnostic features include a fibrous capsule, hyaline vessels, cellular (Antoni A) and loose-textured (Antoni B) areas, and Verocay bodies (opposing rows of spindle nuclei separated by anucleate rows of eosinophilic processes).16 Unlike neurofibromas, schwannomas arise within the nerve sheath and displace the fascicles as opposed to enveloping them (Fig. 203-5). However, often a small fascicle entering or exiting the proximal and distal poles of the tumor can be identified.
Molecular Biology
Mutations in the NF2 gene at the 22q12.2 locus are found in sporadic schwannomas and those in patients with NF2.18 The schwannomas that develop in patients with schwannomatosis show a multitude of truncations in the NF2 gene, and multiple tumors from an individual patient can each have unique mutations.15,19 The NF2 tumor suppressor gene codes for merlin (schwannomin), a cell membrane protein that functions in intercellular signaling and helps mediate growth arrest. The loss of both wild type NF2 alleles (two-hit hypothesis of tumorigenesis) results in marked reduction in merlin protein and tumor development.20
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
 Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


