and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
1.1 Epidemiology of Peripheral Neuropathy
Diseases of the peripheral nerves represent a significant neurological problem, with the incidence of polyneuropathy in the USA estimated at 40 per 100,000, an incidence comparable to that of epilepsy or parkinsonism (Kurtzke 1982), and an overall prevalence of about 2.4 % in the general population (Martyn and Hughes 1997). Because not all patients are investigated, data regarding the etiologic composition of polyneuropathies is difficult to obtain. For example, Dyck et al. estimated that only 10 % of affected patients in kinships with hypertrophic hereditary motor and sensory neuropathy (HMSN-1 or Charcot–Marie–Tooth disease (CMT) type 1) seek medical attention as a direct consequence of symptoms produced by this disease (Dyck et al. 1993). Many individuals with mild or subclinical neuropathy, whether genetically determined or acquired, may not require investigation or treatment. The largest and best-documented peripheral neuropathy series originate from specialized centers or are part of selected biopsy series and thus are not representative of neuropathy in the general population.
1.1.1 Etiologies of Peripheral Neuropathy
It is helpful to classify the major etiologies of peripheral neuropathy into acquired toxic or metabolic, inflammatory or infectious, neoplastic and paraprotein associated, and genetically determined (see Table 8.4 for details). Acquired toxic or metabolic factors probably account for the majority of neuropathies, and common causes include diabetes, alcoholism, nutritional deficiencies, and pharmaceutical neurotoxins. The most frequent inflammatory and infectious neuropathies are Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), vasculitic neuropathies, HIV-associated neuropathies, and leprosy. Malignancy is associated with neuropathy through several mechanisms including paraneoplastic phenomena, metabolic derangement, and direct infiltration of nerve by neoplasm. The most common familial neuropathies are CMT types 1 and 2. In the literature, 10–20 % of neuropathies remain cryptogenic (vide infra). Reported series based on consecutive patients from well-defined geographic areas give some insight into the relative frequency of these neuropathies. Prineas (1970) reviewed 278 consecutive English patients hospitalized with a diagnosis of polyneuropathy between 1957 and 1966, excluding 49 patients with questionable or inadequately investigated neuropathies. Approximately 30–35 % of patients had disorders associated with toxic or metabolic causes, with nutritional polyneuropathy and alcoholism forming the largest group. Smaller groups had vitamin B12 deficiency (7 %), drug-induced neuropathies (7 %), malignancy associated (7 %), and diabetes (5 %). Retrospective examination of the data suggests that GBS accounted for 20–25 % and CIDP 7–15 %. Workup of Mayo Clinic patients (n = 205) with an undiagnosed peripheral neuropathy eventually resulted in a diagnosis in 76 % (Dyck et al. 1981). CIDP was diagnosed in 21 %, hereditary neuropathy in 42 %, and the remainder (diabetes, toxins, and neoplasia) accounted for 13 %. It is likely that the chronic idiopathic group included some genetically determined neuropathies for which a family history was not obtained.
A very large series (>5,000 cases) including autopsy and nerve biopsy examination resulted in specific diagnoses in only 23 % (Schröder 1998); common diagnoses included vasculitis (64 %), Guillain–Barré (10 %), and amyloid (4 %). In a report on 120 consecutive patients with polyneuropathy seen over a 3-year period in an EMG clinic in Israel, diabetic neuropathy accounted for 22 %, while 20 % had GBS, CIDP, or vasculitis; 13 % were genetically determined, 13 % metabolic, 6 % drug induced, 4 % associated with malignancy, 8 % miscellaneous, and 12 % remained undiagnosed (Argov et al. 1989). A 380-patient prospective peripheral neuropathy series from France revealed alcohol or drug toxicity in 23 % of patients, diabetic neuropathies in 16 %, GBS and CIDP in 16 %, hereditary neuropathies in 10 %, and neuropathies associated with collagen diseases in 10 % (Vallat et al. 1983). A large (nearly 1,000 consecutive cases) series of nerve biopsies performed at the University of Toronto established a specific diagnosis in approximately 29 % of cases (Bilbao 2004). Barohn’s (1998) experience of 402 consecutive patients referred to the University of Texas neuromuscular outpatient clinics differed, with the most frequent diagnoses being hereditary (30 %), cryptogenic sensory polyneuropathy (23 %), diabetes (15 %), and inflammatory demyelinating polyneuropathy (13 %). Diabetic neuropathy is consistently underestimated in such studies because many diabetic patients are not referred to neurologists, are rarely admitted to hospital because of neuropathy, or are only biopsied if they have an atypical presentation or course.
Thus, despite the absence of information about the types and frequency of polyneuropathy in the general population, a first estimate suggests that in the developed world metabolic (including diabetes), toxic, and nutritional causes account for 50 % or more of neuropathies, inflammatory neuropathies (mainly GBS, CIDP, and vasculitis) for 10–20 %, familial neuropathy for 10–20 %, and neoplasia-associated neuropathy for 5–10%, and approximately 10–20 % remain idiopathic. A large variety of rare neuropathies make up the remaining cases. In pediatric neuropathy, the etiologic spectrum is different, with familial neuropathies predominating (Ouvrier et al. 1990).
Geographic factors are pertinent in determining the relative frequency of neuropathies in a given population. In a large Indian series, 66 % of neuropathies were diabetic, 14 % leprous, and 11 % GBS (Wadia 1984). Leprous, nutritional, and familial neuropathies were underrepresented and GBS overrepresented in this hospital inpatient series. In a 1980 review, Osuntokun (1980) estimated that in Africa 25–40 % of neuropathies seen by neurologists were nutritional, with 5–10 % each due to leprosy, alcoholism, and the Guillain–Barré syndrome. Diabetes was accorded equal ranking with porphyria at about 3 % each! However, this author noted that when all patients are considered, including those not coming to neurologists’ attention, leprosy was by far the most important cause of neuropathy, and diabetic neuropathies became more numerous. HIV has become an increasingly important cause of peripheral neuropathy worldwide, with an estimated prevalence of between 10 and 60 % (Schifitto et al. 2002).
1.1.2 Cryptogenic Peripheral Neuropathy
Despite widely variable selection criteria, the proportion of neuropathies which remain cryptogenic is remarkably consistent and ranges from 10 to 25 % (Argov et al. 1989; Corvisier et al. 1987; Grahmann et al. 1991; McLeod et al. 1984; Vallat et al. 1983; Wolfe et al. 1999). Series from the 1950s and 1960s reported 56–70 % of polyneuropathies as cryptogenic (Matthews 1952; Rose 1960), but several currently accepted “etiologies” of peripheral neuropathy were not included, such as GBS, CIDP, paraprotein-associated neuropathy, paraneoplastic neuropathy, as well as the notion of genetically determined neuropathies without a family history (Dyck et al. 1981).
When patients with a chronic idiopathic neuropathy are restudied, an etiologic diagnosis can often be made. McLeod et al. (1984) reported that about 1/3 of their patients with chronic idiopathic neuropathies could be subsequently diagnosed at a 1-year interval, and Grahmann et al. (1991) reported similar numbers. In the previously mentioned review of 205 unclassified patients, an etiologic diagnosis was eventually made in 76 % (Dyck et al. 1981); however, this group was probably not as well studied prior to referral as the cryptogenic patients in the series of McLeod et al. (1984). In contrast to these reports, Jann et al. (2001) found that an etiology could not be determined over a 4-year follow-up period for 40 consecutive patients with idiopathic sensorimotor polyneuropathy. Similarly, Notermans et al. (1994) found an etiology in only 4 out of 75 cases of cryptogenic neuropathy over a 5-year follow-up. These authors suggest that in patients with a minimally progressive polyneuropathy of undetermined cause despite extensive workup, regular repeated investigations may not be warranted.
The terminology for cryptogenic neuropathies has evolved, and the current preferred term is chronic idiopathic axonal polyneuropathy (CIAP) (Singer et al. 2012). Recent data suggest that after a detailed assessment, patients ultimately diagnosed with CIAP tend to have a slowly progressive course, with minimal weakness or disability (Jann et al. 2001; Notermans et al. 1994; Wolfe et al. 1999). Patients with CIAP tend to present in their 50s or 60s with an insidious onset of predominantly sensory symptoms of numbness and paresthesias, occurring in a length-dependent manner. Power is relatively preserved, and reflexes are reduced or absent. Indeed, based on consistent clinical features and course, diagnostic criteria have been proposed to identify CIAP as a distinct clinical entity (Wolfe et al. 1999; Singer et al. 2012).
These studies emphasize the importance of careful history-taking in diagnosis, especially for evidence of a familial neuropathy or of toxic exposure (Dyck et al. 1981; Jann et al. 2001; McLeod et al. 1984). Examination of family members, even if not known to be symptomatic, can prove extremely helpful. In other patients, the etiology becomes clear when malignancy or other systemic conditions emerge, and reassessment is necessary if the clinical course deviates from what is expected in CIAP.
1.2 Usefulness of Nerve Biopsy in Evaluation of Peripheral Neuropathy
For several classes of patients, nerve biopsy is rarely necessary: patients with an established cause of toxic or metabolic neuropathy, patients with a familial neuropathy, those with the Guillain–Barré syndrome, or in patients who are known to have a circulating paraprotein or systemic malignancy. In addition, patients with suspected CIDP may not require biopsy (vide infra). Nevertheless, biopsy continues to have a significant role in the diagnosis of peripheral nerve disease (Table 1.1).
Table 1.1
Specific diagnoses that can be made by nerve biopsy
Inflammatory/infectious |
Neuropathy with macrophage-mediated demyelination (CIDP, GBS) |
Vasculitic neuropathy |
Leprous neuropathy, especially primary neuritic leprosy |
Sarcoidosis or granulomatous neuropathy |
CMV neuritis in immunosuppressed patients |
Neoplasm/paraprotein associated |
Non-amyloid paraprotein-associated neuropathies |
IgM anti-MAG paraprotein (widely spaced myelin)a |
POEMS syndrome (uncompacted myelin)a |
Immunoglobulin deposition disease |
Primary amyloidosis |
Neoplastic infiltrative neuropathy |
Lymphomatoid granulomatosis |
Metabolic/toxic |
Amiodarone neuropathy |
Hexacarbon neuropathy |
Genetically determined |
Hereditary neuropathy with liability to pressure palsiesb |
Amyloid neuropathy, familial |
Giant axonal neuropathy |
Neuroaxonal dystrophy |
Polyglucosan body disease |
Hereditary sensory and autonomic neuropathies |
Storage diseases |
Metachromatic leukodystrophyb |
Adrenoleukodystrophyb |
Globoid cell leukodystrophyb |
Niemann–Pick diseaseb |
Fabry diseaseb |
Tangier diseaseb |
Neuronal ceroid lipofuscinosis |
1.2.1 Review of Experience at St. Michael’s Hospital
1.2.1.1 Histological Material and Clinical Data
Since 1972, the peripheral nerve pathology laboratory at St. Michael’s Hospital has processed nearly 700 nerve biopsies. These consist almost exclusively of sural nerves, with infrequent utilization of cutaneous branches from the radial or ulnar nerves, or the lateral peroneal nerve. During this period of time, the same pathologist (JMB) has used a consistent analytic protocol (see Chap. 7) to evaluate all of these cases. The reports of 267 consecutive nerve biopsies examined in the years 1986–1993 were reviewed and the histological changes described classified as either nonspecific (axonal degeneration, segmental demyelination, mixed axonal/demyelinating, and inflammatory or noninflammatory) or findings that permit a specific diagnosis in the absence of clinical information (e.g., vasculitis, amyloid). The clinician most involved in the case used all available information including biopsy findings and follow-up to make the final diagnosis. Based on the available clinical material, the nerve biopsy was classified as being of no value, helpful, or essential for optimal patient management.
A biopsy of no value did not modify management other than providing an impression of the severity and activity of the disease. Helpful biopsies (a) supported the diagnosis of a suspected etiology for which treatment or genetic counseling is available (e.g., CIDP, CMT), (b) ruled out a working diagnosis (e.g., finding of prominent active Wallerian changes in suspected CMT-2), and (c) distinguished between two alternative diagnoses with therapeutic implications (e.g., leprous vs. vasculitic neuropathy in a patient from a leprosy-endemic region). Biopsies essential to management revealed abnormalities that (a) permitted definitive diagnosis of the cause of the neuropathy and (b) revealed suggestive but not completely diagnostic findings, resulting in altered management (e.g., non-caseating granulomata suggesting sarcoidosis or findings typical of CIDP in an “axonal” neuropathy).
1.2.1.2 Results
In a review of nerve biopsies and clinical records for 267 patients with a mean age of 57.5 years (Fig. 1.1), 234 patients had sufficient clinical material to permit accurate clinicopathological correlation. In these 234 patients, a final etiologic diagnosis was reached in 76 % (Table 1.2). Diagnostic findings were seen in 16 % of all nerve biopsies (Table 1.3).
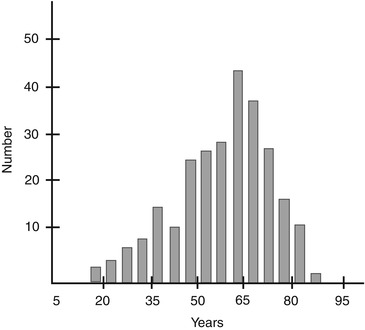
Fig. 1.1
Age distribution of patients for 267 consecutive nerve biopsies at Saint Michael’s Hospital 1986–1993
Table 1.2
Clinicopathological correlations in 267 nerve biopsies
Specific etiology | Nonspecific histology | ||||||
|---|---|---|---|---|---|---|---|
Noninflammatory | Inflammatory | ||||||
Histology | N | Mixed | Axonal | Mixed/demyel | Axonal | Mixed/demyel | Normal |
Final diagnosis obtained (N = 177) | |||||||
CIDP/GBS) | 51 | (4)c | 5 | 12 | 7 | 27 | 1 |
Vasculitis | 19 | 19 | 0 | 0 | 0 | 0 | 0 |
Paraprotein | 12 | 2 | 3 | 3 | 1 | 2 | 1 |
NASID | 11 | 0 | 3 | 1 | 4 | 2 | 1 |
Diabetes | 10 | 0 | 5 | 2 | 2 | 1 | 0 |
Toxic/nutritionala | 10 | 3 | 6 | 1 | 0 | 0 | 0 |
HMSN-1 | 5 | 0 | 0 | 5 | 0 | 0 | 0 |
HMSN-2 | 7 | 0 | 7 | 0 | 0 | 0 | 0 |
Amyloid | 5 | 4 | 1 | 0 | 0 | 0 | 0 |
Uremia | 4 | 0 | 3 | 1 | 0 | 0 | 0 |
Paraneoplastic | 4 | 0 | 2 | 0 | 2 | 0 | 0 |
Othersb | 13 | 10 | 2 | 0 | 1 | 0 | 0 |
Not neuropathy | 25 | 0 | 7 | 3 | 0 | 0 | 15 |
Final diagnosis not obtained (N = 90) | |||||||
Cryptogenic | 52 | 0 | 29 | 10 | 7 | 2 | 4 |
Insufficient data | 33 | 1 | 19 | 2 | 6 | 2 | 3 |
Random | 5 | 0 | 2 | 1 | 0 | 0 | 2 |
Total | 267 | 39 | 94 | 41 | 30 | 36 | 27 |
15 % | 35 % | 15 % | 11 % | 13 % | 10 % | ||
Table 1.3
Specific findings in 43 biopsies
Necrotizing vasculitis | 20 |
Amyloidosis | 4 (2 familial TTR, 2 primary) |
CIDP (MMD)a | 4 |
Tomaculous neuropathy | 3 |
Amiodarone | 3 |
Leprosy | 2 |
Paraproteinb | 2 |
Granulomatous (sarcoid) | 2 |
Lymphoma (infiltrative) | 2 |
Fabry disease | 1 |
Nerve biopsy was interpreted as essential for management in 48 patients (21 %). In most of these cases, a specific diagnosis was made (e.g., vasculitis, amyloidosis, leprosy). On three occasions, biopsy revealed a picture typical of previously unsuspected CIDP. In one patient clinically diagnosed with CMT-1, biopsy revealed unequivocal inflammatory features suggestive of CIDP. In two patients with a clinical diagnosis of CMT-2, biopsy revealed additional pathological findings that led to specific disease-modifying treatment; in one patient a toxic medication was discontinued, and in another, steroids were given.
We considered nerve biopsy to be helpful in 52 patients (22 %). In most of these cases, biopsies supported a diagnosis of CIDP, CMT-1, or CMT-2, in other instances as distinguishing between paraneoplastic and chemotherapy-related neuropathy by showing prominent inflammation or confirming the diagnosis of GBS in a patient with atypical history and physical findings. In one patient, nerve biopsy revealed selective small myelinated fiber loss suggestive of amyloidosis, but no amyloid was identified; a persistent search in rectal and muscle tissue indeed confirmed this suspicion.
The 267 consecutive patients studied were identified in the pathology laboratory and thus biased towards diagnostically challenging neuropathies and towards etiologies for which a biopsy is used to confirm a suspected treatable diagnosis. This circumstance presumably accounts for the overrepresentation of CIDP and vasculitis in the final diagnoses and for the relatively high rate of cryptogenic neuropathies (24 %) (Table 1.2). Approximately half of the patients with cryptogenic neuropathy suffered from a minimally disabling and nonprogressive or slowly progressive neuropathy, as has been previously documented (Jann et al. 2001; Wolfe et al. 1999; Grahmann 1991; McLeod et al. 1984).
Analysis of this material suggests that no difference exists in the diagnostic value of nerve biopsy between neuropathies with axonal and demyelinating or mixed electrophysiological features (Table 1.4). Indeed, nerve biopsies in patients with axonal neuropathy reveal important and often unsuspected pathology such as vasculitis and amyloidosis. In addition, biopsies in three patients with axonal electrophysiology showed a picture typical of CIDP.
Table 1.4
Usefulness of biopsy according to results of electrophysiological studies
Axonal | Demyelination/mixed | Normal | |
|---|---|---|---|
No benefit | 64 | 21 | 31 |
Helpful | 21 (19 %) | 30 (46 %) | 0 |
Essential | 24 (22 %) | 14 (22 %) | 2 (6 %) |
1.2.2 Literature Data on the Usefulness of Nerve Biopsy
Fifty-three of one hundred and twenty consecutive patients presenting to an EMG clinic underwent nerve biopsy (Argov et al. 1989). Thirty-eight percent of the 53 biopsies yielded diagnostic or contributory information that altered management in half (19 %) of the patients. In another series of 56 sural nerve biopsies, 27 % provided information considered crucial for diagnosis. In an additional 37 %, biopsy findings were nonspecific but supported the suspected clinical diagnosis and excluded elements of the differential diagnosis (Neundorfer et al. 1990). Reviewing personal experience with 385 biopsies, Oh (1990) was able to make a specific diagnosis in 24 % of the cases. Chia et al. (1996) found a high incidence of vasculitis in nerve biopsies (one-third of 100 biopsies) of elderly patients with disabling neuropathy, concluding that nerve biopsy should be considered in all elderly patients with multifocal neuropathies of unknown cause. In another study Deprez et al. (2000) analyzed the clinical features that seemed to increase the diagnostic value of nerve biopsy. These authors found that the biopsy yielded helpful or essential information in 50–60 % of cases with a pre-biopsy diagnosis of vasculitis or CIDP. Additional features that have been shown to improve the usefulness of nerve biopsy include multifocal clinical or electrophysiological findings and performance of the biopsy within 6 months of symptom onset (Schweikert et al. 2007; Deprez et al. 2000). Finally, in a series of 50 consecutive cases, sural nerve biopsy changed the diagnosis in 14 %, altered management in 60 %, and caused persistent increased pain after short-term follow-up in 33 % (Gabriel et al. 2000).
Thus overall, 20–30 % of nerve biopsies provide information that is either critical to diagnosis or may alter management. Nerve biopsy yield is higher if the neuropathy is acute, subacute, or multifocal (i.e., the clinical setting in which the suspicion for a vasculitic neuropathy is high).
1.3 Indications for Nerve Biopsy
Indications for nerve biopsy have not been delineated. A 1967 position paper from the Research Group in Neuromuscular Diseases of the World Federation of Neurology indicated that “…nerve biopsy is an investigation which should only be carried out in selected centers where facilities for full and detailed histological studies are available, and the clinical indications for the use of this method of investigation are few” (Thomas 1970). Although we find this a restrictive statement, we agree that interpretation of pathological change in a nerve specimen requires experience. Many problems that beset the beginner relate to artifacts produced by the biopsy procedure (crush, stretch), during processing (rough handling, delayed fixation, hyperosmolar fixative), and embedding (lack of orientation of fascicles, type of resin). However, the techniques necessary for the processing of nerve tissue can easily be adapted from those found in any established histology laboratory; this includes plastic resin embedding, electron microscopy, and immunohistochemistry in paraffin, frozen, and plastic sections. Diagnosis rarely requires the more difficult technique of nerve teasing. With respect to the indications for nerve biopsy, the position quoted above was taken before (A) full appreciation of CIDP as a treatable neuropathy, (B) the establishment of immunostaining techniques (e.g., identification of amyloid major proteins), and (C) the emergence of such entities as nonsystemic peripheral vasculitic neuropathy (Dyck et al. 1987), giant axonal neuropathy (Asbury et al. 1972), and hereditary neuropathy with liability to pressure palsies (HNPP) (Behse et al. 1972). Furthermore, using plastic-embedded material for light and electron microscopy permits a far more informative examination of axons and myelin than do the standard paraffin-based histological techniques employed in the 1960s and earlier.
Asbury and Gilliatt (1984) emphasized the usefulness of nerve biopsy in asymmetric and multifocal neuropathies and indicated that “… distal symmetrical polyneuropathies, whether subacute or chronic, axonal or demyelinating, are not further clarified by nerve biopsy….” However, in our experience a significantly asymmetric neuropathy occurred in only a third of the instances in which biopsy was classified as essential for management. Significantly, asymmetric neuropathy appeared in approximately 50–70 % of vasculitic neuropathies and in HNPP. A symmetrical and distally predominant pattern was seen in a significant proportion of patients with vasculitis and in all of the patients with amyloid, sarcoid, amiodarone-related, infiltrative, paraprotein-associated, and inflammatory demyelinating neuropathies.
The spectrum and frequency of findings in our series are very similar to those reported in the literature (Argov et al. 1989; Neundorfer et al. 1990; Oh 1990). This suggests that clinicians in widely separated institutions apply similar indications for nerve biopsy despite the absence of accepted guidelines for performing the procedure. As noted above, biopsy reveals essential information in about 20 % of cases and helpful information in a further 20 %. Clearly, nerve biopsy can be a valuable procedure. Unfortunately, 50 % or more of nerve biopsies in these reports have not provided any useful information. Identifying a group of patients who should not undergo nerve biopsy would lower the frequency of uninformative procedures. Although some workers refer to “appropriate selection” of patients for biopsy, there are no concrete guidelines (England and Asbury 2004; Said 1999).
1.3.1 Clinical and Electrophysiological Criteria Cannot Exclude the Need for Biopsy
Although some workers have suggested that axonal neuropathies have a lower diagnostic yield than demyelinating ones (Argov et al. 1989), our experience (Table 1.4), and that of Neundorfer et al. (1990), does not support this assertion. Asbury and Gilliatt (1984) have commented on the low yield of nerve biopsy in distal symmetrical neuropathies, but we have identified vasculitis, amyloidosis, granulomata, and paraprotein deposition in this context.
Most importantly, clinical criteria cannot exclude the possibility of vasculitic neuropathy. Vasculitis was seen in 7 % of our biopsies and in 4–16 % of other literature series (Schweikert et al. 2007; Harati and Niakan 1986; Hawke et al. 1991; Oh 1990). In a group of 100 elderly patients with a disabling neuropathy severe enough to justify biopsy, 33 % were found to have a vasculitic neuropathy, and 30 % of those had a distal symmetrical or distal mildly asymmetric presentation (Chia et al. 1996). Our experience with 32 consecutive biopsy-proven cases where clinical information was available suggests that the most common syndrome seen in vasculitic neuropathy is a distal symmetrical polyneuropathy (46 %). Thirty-eight percent of patients demonstrated mononeuropathy multiplex, and 16 % had an asymmetric polyneuropathy. Notwithstanding the fact that biopsy is more likely to be performed in patients with mononeuropathy multiplex, an acute, subacute, or chronically progressive symmetrical distal sensorimotor polyneuropathy, where vasculitis is not suspected, has been described in 19–76 % of nerve biopsy series. Furthermore, although the classical electrophysiological finding of vasculitis is axonal neuropathy, 3 of 32 patients with necrotizing vasculitis proven by examination of nerve biopsy in St. Michael’s Hospital laboratory demonstrated significant conduction velocity slowing or conduction block. Finally, vasculitis may be confined to the peripheral nervous system, as occurred in 4 of our 32 patients. Consequently, in some instances nerve biopsy alone can identify this treatable cause of neuropathy.
The low diagnostic yield (31 of 33 biopsies were of no value) of biopsy in patients with normal nerve conduction studies (NCS) is not surprising. Most of these patients were ultimately found not to have a neuropathy. However, in two instances specific (and in one case completely unexpected) diagnoses were made: Fabry disease and a granulomatous neuropathy in a patient shown to have sarcoidosis only at autopsy 5 years later.
1.3.2 Nerve Biopsy Has Limited Usefulness in Suspected CIDP
In the years 1986–1993, the single most common indication for nerve biopsy in Saint Michael’s Hospital laboratory was the clinicians’ desire to rule in or out the diagnosis of CIDP. Workers in other laboratories report a similar experience (Solders 1988). However, as discussed in Chap. 9, our data and the literature suggest that biopsy cannot exclude the diagnosis of CIDP unless an alternative specific diagnosis is made. In one study, sural nerve biopsy failed to show significant pathological differences between cases of CIDP and chronic axonal polyneuropathy (Bosboom et al. 2001). Thus, there may be little value in performing a biopsy for suspected CIDP (Krendel et al. 1989). Nerve biopsy can distinguish between CIDP and CMT-1 or identify CMT-1 with superimposed CIDP, but clinical and electrophysiological criteria, as well as genetic testing, usually suffice (Dyck et al. 1982; Gabreels-Festen et al. 1993; Sladky et al. 1986).
More important than confirming already suspected CIDP is detection of this condition in the absence of clinical evidence. Histological findings typical of CIDP can be seen in electrophysiological axonal neuropathies (Barohn et al. 1989), as occurred in 3 of 109 patients in our series, thus permitting effective treatment.
1.3.3 Suggested Guidelines for Use of Nerve Biopsy
One cannot set rigid criteria for the indications of nerve biopsy. If the clinician cannot arrive at a diagnosis after all clinical and electrophysiological tests are exhausted, including screening for inherited neuropathy in the patient’s relatives, the neuropathy is considered to be cryptogenic, as discussed above. Patients with mild and slowly progressive cryptogenic polyneuropathy should undergo a period of observation and repeat evaluation (Jann et al. 2001; Notermans et al. 1994; Grahmann et al. 1981; McLeod et al. 1984). We have also had the experience of making a definite diagnosis of a treatable condition on biopsy in cases where the patients did not subsequently receive treatment. A patient may refuse treatment because of the mildness of the neuropathy, because of the toxicity of the therapy (usually steroid treatment), or because the patient was at the end stage of a severe illness (e.g., HIV).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


