Chapter 38 Peroxisomal Disorders
Disorders of the peroxisome – organelles found in all eukaryotic cells – are characterized by alterations in their unique metabolic functions in the cell and tissues. The pervasive presence of the peroxisome leads to far-reaching consequences of these genetic disorders. Peroxisomal disorders are divided into two major categories. In the first, the organelle fails to develop normally, leading to disruption of multiple peroxisomal enzymes. The second category consists of those disorders in which the peroxisome structure is normal but functioning of a single peroxisomal enzyme is defective. Box 38-1 lists the known peroxisomal disorders; their combined incidence is estimated at 1 in 25,000 or higher [Heymans, 1984]. Because peroxisomal disorders are genetically determined, with a majority readily identifiable by biochemical means, including prenatal testing, and nearly all affecting the nervous system, knowledge of these diseases is important. As the pathophysiology of these disorders is better understood, novel therapeutic strategies may be developed, which gives new hope for advances not only in the diagnosis but also in the management and outcome of peroxisomal disorders.
Box 38-1 Peroxisomal Disorders













CADDS, contiguous ABCD1 DXS1357E deletion syndrome; CoA, coenzyme A; DHAP, dihydroxyacetone phosphate.
Historical Overview
The organelle was identified in 1954 by Rhodin and named the “microbody” [Rhodin, 1954]. In 1960, it was found that this structure contained urate, d-amino acid oxidase, and catalase; it was named the peroxisome by de Duve and Baudhuin [1966]. Recognition of its function in active cellular processes came with the demonstration of its role in the β-oxidation of fatty acids (Figure 38-1) [Lazarow, 1978] and plasmalogen synthesis [Hajra and Bishop, 1982]. That change in peroxisome function results in disease was not evident until 1973, when Goldfischer and colleagues determined its absence in Zellweger’s syndrome [Goldfischer et al., 1973]. The link between peroxisomes and human disease was not made until that time, even though peroxisomal disorders had been described much earlier, including X-linked adrenoleukodystrophy in 1923 [Siemerling and Creutzfeldt, 1923], acatalasemia in 1948 [Takahara and Migamoto, 1948], and Zellweger’s syndrome in 1964 [Bowen et al., 1964]. From the initial three enzymes identified, more than 40 enzymes have been localized to the peroxisomes [Tolbert, 1981].

Structure and Function of Peroxisomes
The peroxisome is bound by a single membrane and contains a fine granular matrix. Histologically, these organelles are identified by the presence of catalase, are present in all human tissues except mature erythrocytes, and demonstrate variation in size and number. In liver and kidney, where they are abundant, the average diameter is 500 nm, whereas in the nervous system, fibroblasts, and amniocytes, they measure 100–250 nm [Hruban and Rechcigl, 1969]. They do not contain DNA and appear to be devoid of glycoproteins. The membrane is 6.5–7 nm thick and has a trilaminar appearance and a unique protein composition. Peroxisome membrane proteins with molecular masses of 22, 26, 27, 41, 57, 68, and 70 kDa have been identified [Hashimoto et al., 1986; Imanaka et al., 1991]. The peroxisomal membrane also contains four ATP binding cassette (ABC) proteins. Although the precise role of these proteins in the peroxisomal membrane is still under investigation, it is known that this family of proteins has important intracellular roles in transport and other functions. As discussed later on, the ABC protein, ABCD1, is defective in X-linked adrenoleukodystrophy, the most prevalent peroxisomal disorder [Wanders et al., 2007].
The process of peroxisomal biogenesis is highly conserved in all eukaryotic organisms, which has permitted the study of yeast to identify the cellular mechanism for the assembly of the organelle and targeting of proteins to the developing vesicle [Weller et al., 2003]. The early concept of peroxisome biogenesis by budding of the endoplasmic reticulum has undergone significant revision. Peroxisomal proteins are encoded by nuclear genes, synthesized on free polyribosomes, and discharged into the cytosol in the mature form. Work in yeast has identified more than 20 genes labeled PEX whose products, peroxins, are required for the incorporation of peroxisome membrane proteins and matrix protein importation. Peroxins are required for the proper importation and have roles in receptor docking, stability, and translocation across the membrane [Weller et al., 2003; Ma and Subramani, 2009].
Targeting information directing matrix proteins into the peroxisomes is inherent in the mature polypeptide. A majority of proteins destined for the peroxisome use peroxisome targeting sequence 1 (PTS1), which consists of a terminal tripeptide of serine-lysine-leucine (-SKL) that is recognized by the soluble receptor Pex5p [Gould et al., 1987, 1988, 1990; Keller et al., 1991; Miyazawa et al., 1989]. Not all matrix proteins contain the carboxyl-terminal PTS1 signal. Peroxisomal 3-ketoacyl-coenzyme A (CoA) thiolase and phytanoyl-CoA hydroxylase have a different peroxisomal targeting sequence (PTS2). PTS2 consists of a nine-residue signal located at the amino terminus. It directs the import of a smaller number of proteins using the soluble receptor Pex7p [Swinkels et al., 1991].
Both Pex5p and Pex7p receptors bind their targeted proteins in the cytoplasm outside the peroxisome. The present model has the receptor-peptide complex dock at the peroxisome surface by means of membrane-associated complexes containing other peroxisome assembly proteins including Pex3p, Pex13p, Pex14p, and Pex17p. Other peroxins appear to function later in the process of translocation. Several are zinc-binding proteins acting downstream of the docking complex and are postulated to constitute the translocation complex involved in matrix protein import. Recent evidence has demonstrated that Pex5p is translocated into the peroxisome and is capable of returning to the cytoplasm – an extended shuttle model [Dammai and Subramani, 2001].
This import method has no parallel with any other organelle. Walton and colleagues demonstrated that peroxisome import allows the uptake of folded, oligomerized proteins and even the import of non-PTS-containing substances, so long as it was with PTS-containing cargo [Walton et al., 1995]. This mechanism is in contrast with that in the mitochondria and lysosome, which have a translocon mechanism with structural modification occurring within the targeted organelle [Lanyon-Hogg et al., 2010].
In contrast with the proteins targeted for the matrix, the proteins used in the membrane utilize another mechanism. Jones and associates examined PMP34 and found that it contained at least two targeting regions. Examining another peroxisomal membrane protein, Pex13, these investigators again found that it had multiple nonoverlapping targeting signals [Jones et al., 2001]. These and other peroxisomal membrane proteins did not share targeting regions. Regions were relatively long and contained at least one membrane-spanning domain.
Peroxisomal number and division appear to be under metabolic control. Hypolipidemic agents, such as clofibrate and industrial phthalate plasticizers, result in the proliferation of liver peroxisomes [Hess et al., 1965; Lazarow et al., 1985]. These agents induce a 20- to 30-fold increase in the activity of the fatty acid β-oxidation system by the rapid coordinated increase of peroxisomal acyl coenzyme A (acyl-CoA) oxidase and bifunctional enzyme [Reddy et al., 1986]. This effect is mediated by activation of peroxisome proliferator-activated receptorα (PPARα), a binding protein related to the steroid hormone receptor superfamily [Issemann and Green, 1990; Lalwani et al., 1987]. Activated PPARα heterodimerizes with a second member of the nuclear receptor superfamily, retinoid X receptor, to form an active transcription factor [Weller et al., 2003]. Peroxisomes also are induced by high-fat diets [Neat et al., 1980], by adrenocorticotropic hormone [Black and Russo, 1980], by thyroid hormone [Fringes and Reith, 1982], and by the agent 4-phenylbutyrate [Wei et al., 2000]. Peroxisomal function is therefore influenced by metabolic state, nutrition, and pharmaceutical agents.
Reflecting this genetic and metabolic control, peroxisomes vary in tissues throughout the body and during development. In the nervous system of the rat, the peroxisomes are smaller, approximately 140 nm, and fewer than in the liver. They predominate in the first 2 weeks of life in the glia and neurons of the cerebrum, cerebellum, locus coeruleus, and spinal cord, but are fewer in the neurons of adult animals [Nagase et al., 2004]. They are rare in oligodendrocytes in neonatal or adult animals, but become prominent during myelin formation, and may lie adjacent to the outer lamellae that form the myelin sheath [Arnold and Holtzman, 1978]. The biochemical activities follow a similar pattern wherein the activities of catalase and peroxisomal acyl-CoA oxidase and oxidation of lignoceric acid in brain reach a peak at postnatal days 10–16 and then decline. This decline suggests that peroxisomes play a vital role during brain development and myelinogenesis, which may account for the severe brain abnormalities noted in the neonatal and infantile forms of these disorders, including neuronal migration defects.
Metabolic Function of Peroxisomes
Peroxisomes were named for the presence of hydrogen peroxide and catalase, which decomposes the hydrogen peroxide. The seminal observation by Lazarow and de Duve in 1976 that rat liver peroxisomes oxidized palmityl-CoA, followed in 1978 by the demonstration of enzymes of β-oxidation and production of acetyl-CoA by Lazarow and Fujiki [1985], led to the recognition of the importance of this organelle in biologic processes. It is now known that more than 40 enzymatic functions are found in the peroxisome. Some peroxisomal activities, such as oxidation of fatty acids and cholesterol synthesis, can occur in other cellular compartments as well. Certain reactions, however, occur exclusively in the peroxisome. These reactions include oxidation of very long chain fatty acids and pipecolic acid and certain steps in the synthesis of plasmalogens and bile acids. These reactions are abnormal in many peroxisomal disorders. The composition of enzymes within the peroxisome varies among species and within tissues in a species, as well as with maturation, metabolic state, and environmental factors. The following discussion focuses on those metabolic pathways that are unique and are used as markers in diagnosis.
Peroxisomal Fatty Acid Oxidation
Peroxisomal β-oxidation enzymes are distinct from their mitochondrial counterparts. The biochemical steps are similar, involving several distinct enzymes including an oxidase, multifunctional enzyme (displaying enoyl-CoA hydratase and 3-hydroxy acyl-CoA dehydrogenase activity), and 3-oxo acyl-CoA thiolase. It recently has become clear that several distinct oxidases, multifunctional proteins, and enzymes catalyzing the thiolytic cleavage exist. The two pathways, l– and d-specific, are depicted in Figure 38-1.
Long-chain acyl-coenzyme a ligase and lignoceroyl-coenzyme a ligase
Initially it was thought that a single acyl-CoA ligase that activated long-chain (C10–C18) fatty acids was involved in oxidation of fatty acids and that it was common to mitochondria, microsomes, and peroxisomes [Miyazawa et al., 1985]. This enzyme is localized to the cytoplasmic side of the peroxisomal membrane, unlike other peroxisomal enzymes, which are in the matrix [Mannaerts et al., 1982]. Subsequently, a series of investigations led to the recognition of a closely related but separate enzyme, which is a ligase for very long chain fatty acids and is referred to as lignoceryl-CoA ligase or synthetase [Singh et al., 1988]. This enzyme is active toward lignoceric acid (C24:0) and hexacosanoic acid (C26:0). Unlike long-chain ligase, very long chain fatty acid ligase is absent in mitochondria. Singh et al. [1984] demonstrated that lignoceric acid is oxidized exclusively in the peroxisome. It is unclear if the very long chain fatty acid ligase plays any role in the pathogenesis of peroxisomal disorders.
Acyl-coenzyme a oxidases
The straight-chain acyl-CoA oxidase is synthesized in the mature form containing the carboxyl-terminal PTS1 [Miyazawa et al., 1987]. The oxidase is most active toward saturated and unsaturated fatty acids with 12- to 18-carbon chain lengths. The oxidase for bile acid intermediates and branched-chain fatty acids is a separate enzyme in humans [Casteels et al., 1990; Ferdinandusse et al., 2003; Scheperse et al., 1990].
Bifunctional or multifunctional enzymes
In both peroxisomal β-oxidation systems, the hydration step (enoyl-CoA hydratase) and the dehydrogenation step (3-hydroxyacyl-CoA dehydrogenase) are catalyzed by multifunctional enzymes (MFEs). The originally identified “bifunctional enzyme,” known as either MFE1 or l-bifunctional protein (l-BP), is l-specific. Human deficiency of the d-specific enzyme, referred to as either MFE2 or d-bifunctional protein (d-BP), causes a failure in the oxidation of very long chain fatty acids and branched-chain fatty acids. MFE2 was found to be identical to 17-hydroxysteroid dehydrogenase type 4 and has a domain at its carboxyl terminus that resembles sterol carrier protein 2 (SCP2), an important intracellular sterol and lipid-binding and transport protein [van Grunsven et al., 1999].
Thiolases
3-Ketoacyl-CoA thiolase is the enzyme for the l-specific pathway and was characterized and cloned first [Bout et al., 1988; Hashimoto, 1982]. Unlike other peroxisomal enzymes but similar to mitochondrial enzymes, it is synthesized as a precursor and converted to the mature form by cleavage of a leader sequence. The peroxisomal targeting sequence for thiolase is located at the amino-terminal end [Swinkels et al., 1991] and was the first protein in which the PTS2 signal was characterized. The thiolytic cleavage in the d-specific pathways is catalyzed by sterol carrier protein X (SCPX). This 58-kDa protein has an amino-terminal thiolase domain that is a phylogenetic equivalent of the mitochondrial and peroxisomal thiolases [Igual et al., 1992].
Oxidation of Unsaturated Fatty Acids
Degradation of monounsaturated and polyunsaturated fatty acids has been demonstrated to occur in peroxisomes, in addition to chain shortening of long-chain dicarboxylic acids [Ferdinandusse et al., 2001; Hiltunen et al., 1986; Kolvraa and Gregersen, 1983; Schulz and Kunau, 1987]. An example of a polyunsaturated fatty acid that requires peroxisomal oxidation is docosahexaenoic acid, which has an important role in retina and brain development, and is known to be deficient in certain peroxisomal disorders [Moser et al., 1999; Watkins et al., 2001].
Branched-Chain Fatty Acid Oxidation and Phytanic Acid α-Oxidation
Branched-chain fatty acids are channeled into the d-specific pathway. The relevant enzymes include branched-chain acyl-CoA oxidase, MFE2, and sterol carrier protein, which functions as thiolase (see Figure 38-1). Phytanic acid has a β-methyl group that blocks β-oxidation and therefore must be α-oxidized first. After activation to its CoA derivative, phytanic acid is α-hydroxylated by phytanoyl CoA α-hydroxylase, the enzyme defective in classic Refsum’s disease [Jansen et al., 1997; Mihalik et al., 1997]. The α-hydroxy phytanoyl CoA is then degraded by a lyase to pristanic acid and formate [Croes et al., 1997]. The pristanic acid then can enter β-oxidation through the branched-chain pathway.
Bile Acid Synthesis
Cholesterol is converted in the liver to cholic acid and chenodeoxycholic acid by a series of enzymatic steps localized to several subcellular compartments. Pedersen and Gustafsson [1980] first demonstrated that the peroxisomal fraction of rat liver catalyzes the conversion of an intermediate product of this pathway, tri- or tetrahydroxy-5-β-cholestanoic acid (trihydroxycholestanoic acid), to cholic acid. Subsequently, Kase et al. [1986] reported that the conversion of trihydroxycholestanoic acid to cholic acid, and of dihydrocholestanoic acid to chenodeoxycholic acid also took place in peroxisomes and was deficient in patients with Zellweger’s syndrome. Shortening of the cholesterol side chain by β-oxidation resulting in the formation of bile acids is analogous to fatty acid oxidation and uses the enzymes of the d-specific peroxisomal β-oxidation pathway. The oxidase for trihydroxycholestanoic acid–CoA is separate from that of fatty acids and in humans also is believed to function as a branched-chain fatty acid oxidase (see Figure 38-1). A cytochrome P-450 catalyzing the hydroxylation of the C26/27 carbon of cholesterol in bile acid synthesis and formation of taurine conjugates of bile acid also has been localized to peroxisomes [Gutierrez et al., 1988; Kase and Bjorkheim, 1989]. In peroxisomal disorders, an increase in plasma and urine bile acid intermediates can be used as a diagnostic criterion.
Plasmalogen Synthesis
Plasmalogens are ether phospholipids and constitute 5–20 percent of phospholipids in mammalian cell membranes [Snyder, 1972]. They are abundant in myelin, in which they constitute one-third of the myelin phospholipids [Norton and Autilio, 1966]. Although the role of ether phospholipids is not fully elucidated, they appear to have important roles as antioxidants in membrane dynamics, storage of polyunsaturated fatty acids, and signal transduction [Brites et al., 2004].
Plasmalogen synthesis is initiated by the acylation of dihydroxyacetone phosphate by dihydroxyacetone phosphate acyltransferase. Experimental studies in cultured skin fibroblasts demonstrate that ATP is required to overcome the latency of dihydroxyacetone phosphate acyltransferase, a finding that is compatible with an ATP-linked translocation of the substrate across the peroxisomal membrane [Wolvetang et al., 1990]. The second enzyme, alkyl-dihydroxyacetone phosphate synthase, replaces the acyl group with a long-chain alcohol [Hajra and Bishop, 1982]. Both enzymes involved in these initial steps of introducing the ether bond into ether phospholipids are located on the inner surface of peroxisome membranes. The third step is the reduction of the alkyl-dihydroxyacetone phosphate to 1-alkyl glycerol-3-phosphate by the enzyme acyl/alkyl-dihydroxyacetone phosphate reductase, which is present in both peroxisomes and microsomes [Datta et al., 1990]. All subsequent steps of synthesis occur in the microsomes. A method developed by Roscher et al. [1985] determines the ratio of the activities in peroxisomal and microsomal components, and has proved to be of value in the study of peroxisomal disorders, many of which feature a marked reduction in plasmalogen synthesis.
Prostaglandin Degradation
Peroxisomes are involved in the chain shortening of prostaglandins F2a [Diczfalusy and Alexson, 1988] and E2 [Schepers et al., 1986]. Tiffany et al. [1991] demonstrated that basal and interleukin-1-stimulated synthesis of prostaglandin E2 increased in a group of patients with X-linked adrenoleukodystrophy.
Amino Acid Metabolism
D-Amino acid oxidase
d-Amino acid oxidase is a flavoprotein with stereospecific activity toward d-amino acids and greatest activity toward d-proline. The kidney has the highest level of activity, although the enzyme is widely distributed in vertebral tissues. The role of d-amino acids as neurotransmitters has recently attracted attention [Errico et al., 2009].
Alanine-glyoxalate aminotransferase
Alanine-glyoxalate aminotransferase is a pyridoxal phosphate-dependent enzyme that catalyzes the transamination of glyoxalate to glycine, with alanine serving as the amino group donor. The subcellular localization of alanine-glyoxalate aminotransferase is species-dependent; it is present in peroxisomes in humans, rabbits, guinea pigs, and macaques, whereas it is mitochondrial in cats and dogs. It is present in both organelles in the rat, mouse, and hamster. The complementary DNA (cDNA) sequence of alanine-glyoxalate aminotransferase has been determined, and the subcellular localization appears to be determined by the targeting signal at the amino terminus [Danpure, 1995; Noguchi and Takada, 1979; Noguchi et al., 1978]. Of great interest is the observation that the mitochondrial targeting signal active in the rat enzyme is not expressed in humans. In some patients with hyperoxaluria type 1, this is altered, resulting in targeting of the alanine-glyoxalate aminotransferase to mitochondria instead of to the peroxisomes [Purdue et al., 1990]. In hyperoxaluria, the defective alanine-glyoxalate aminotransferase results in the conversion of glyoxalate to oxalic acid, which causes oxaluria and nephrocalcinosis.
Pipecolic acid oxidase
l-Pipecolic acid is a component of an alternate lysine degradation pathway and is oxidized by l-pipecolic acid oxidase to α-aminoadipic acid. This enzyme contains flavin, and its subcellular localization is species-dependent. The major site of oxidation is peroxisomal in humans and monkeys, and mitochondrial in rabbits [Mihalik and Rhead, 1989]. The enzyme has been identified in human liver and has been reported to be deficient in Zellweger’s syndrome [Mihalik et al., 1989; Wanders et al., 1988a]. The human enzyme most closely resembles the bacterial monomeric sarcosine oxidases [Dodt et al., 2000]. Isolated increases in pipecolic acid have been reported in normal adults and may be a benign trait [Vallat et al., 1996]. Pipecolic acid elevation has also been seen in pyridoxine-responsive seizures, but this is due not to defects in the oxidase, but rather to the antiquitin (ALDH7A1) gene, which affects other aspects of the pathway [Bennett et al., 2009].
Classification of Peroxisomal Disorders
Peroxisomal disorders may be divided into two categories:
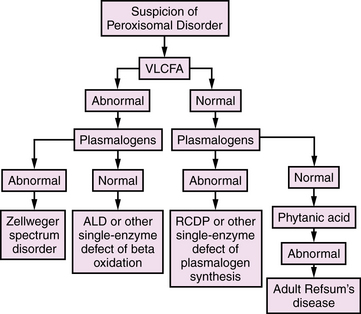
All of these disorders are, in actuality, single-gene and ultimately single-protein deficiencies, but the downstream consequences of the first group affect more than one peroxisomal pathway, resulting in multiple diagnostic abnormalities. With the emergence of DNA-based diagnosis, the utility of this division may require reassessment. An overview of diagnostic evaluation of peroxisomal disorders is provided in Figure 38-2.
Conditions Resulting from Defective Peroxisome Biogenesis
Conditions resulting from defective peroxisome biogenesis are listed in Box 38-1; Zellweger’s syndrome and rhizomelic chondrodysplasia punctata are the respective prototypes. Clinical and biochemical variation between these two types of assembly defects still makes it useful to discuss them separately, although it is important to recognize that these disorders were described on a clinical basis before details of the cell and molecular biology of peroxisomal disorders was known, and hence they were assigned names based on the clinical features, pathologic findings, or biochemical defects that identified them.
Molecular Etiology of Disorders of Peroxisome Assembly
Cell complementation studies were instrumental in the early understanding of the genetics of peroxisomal assembly disorders. Identification of complementation groups provided cell lines that are homogeneous with respect to their gene defect. In conjunction with the use of other molecular biology tools, including the study of yeast and other cell types and the emergence of computerized genomic databases, a new understanding of the molecular underpinnings of the peroxisome assembly disorders has been attained [Lazarow, 1995]. These disorders result from defects in the PEX genes. A complex interaction of peroxins is necessary for the biogenesis of peroxisomes, and a defect in any of these proteins impairs the process. The final common pathway is peroxisomal dysfunction, with the respective clinical syndromes.
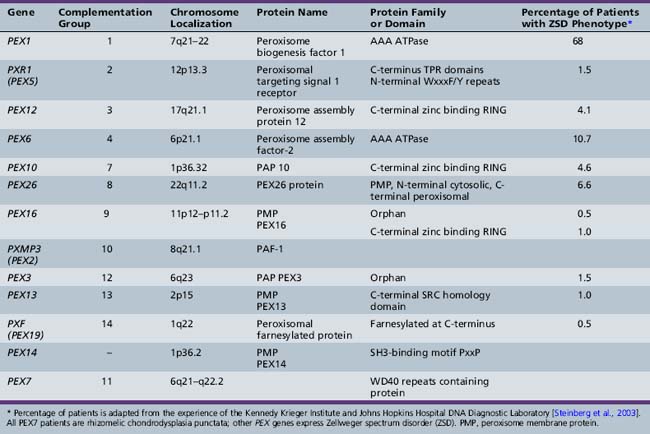
From the study of complementation groups, it was determined that peroxisomal disorders comprise up to 16 complementation groups [Moser et al., 1995]. A majority of the identified groups included persons with Zellweger’s syndrome and milder variations of that condition. One complementation group, however, contained all of the patients with rhizomelic chondrodysplasia punctata, and no overlap between this clinical group and the others was identified. It has subsequently been determined that Zellweger spectrum disorders are secondary to PTS1-mediated pathways including PEX5, and that rhizomelic chondrodysplasia punctata is secondary to mutations in PEX7, the receptor for PTS2 proteins.
The most common causes of Zellweger spectrum disorders are mutations in either PEX1 or PEX6, although to date, 13 PEX genes have been identified as potential causes of these disorders (Table 38-1). PEX1 and PEX6 encode AAA ATPases. Their exact role in matrix protein importation is not certain, but it is evident that they interact. Overexpression of PEX6 or PEX1 can correct mild deficiencies of the other [Germain-Lee et al., 1997; Reuber et al., 1997]. PEX1 defects account for more than 65 percent of cases of such defects [Steinberg et al., 2003; Weller et al., 2003]. A degree of phenotype correlation with the causative mutation has been noted. A single base-pair deletion results in a severe phenotype, and G843D allows a milder phenotype [Collins and Gould, 1999]. Because it is known that PEX1 and PEX6 interact, the mutation G843D affects this interaction [Weller et al., 2003].
Zellweger Spectrum Disorders
Zellweger’s syndrome (cerebrohepatorenal syndrome) was first described by Bowen and associates [Bowen et al., 1964; Opitz, 1985]. Subsequently, the disorders neonatal adrenoleukodystrophy and infantile Refsum’s disease were described as separate entities. Although classic Zellweger’s syndrome is the most severe form with a characteristic phenotype, clinical overlap exists between it and the other forms. All of the Zellweger spectrum disorders share morphologic and biochemical abnormalities, and genetic understanding of the underlying mutations has been obtained in many cases. In view of this overlap, it appears prudent at this time to retain a portion of this clinical nomenclature and refer to the group as Zellweger spectrum disorders.
Clinical and Pathologic Features
Zellweger’s syndrome
Zellweger’s syndrome is a multiple congenital anomaly syndrome characterized by craniofacial abnormalities, eye abnormalities, neuronal migration defects, hepatomegaly, chondrodysplasia punctata, and near-complete absence of peroxisomes. The craniofacial features include a high forehead, hypoplastic supraorbital ridges, epicanthal folds, midface hypoplasia, and a large fontanel (Figure 38-3). The head circumference usually is normal. Reported ocular abnormalities include cataracts, glaucoma, corneal clouding, Brushfield spots, optic nerve hypoplasia, and pigmentary retinal abnormalities. Severe weakness and hypotonia manifest in the newborn period, often accompanied by seizures and apnea [Heymans, 1984; Wilson et al., 1986]. Most affected infants have oromotor dysfunction and require tube feeding. Little psychomotor development ensues, and the average life span is limited, with most affected children surviving for 12–24 months. The facial appearance, Brushfield spots, and profound hypotonia may lead to a consideration of Down syndrome, although the chromosomal determination will eliminate that as a consideration.
Striking abnormalities of neuronal migration unique to Zellweger’s syndrome are evident in the cerebral hemispheres as areas of pachygyria or polymicrogyria localized to the opercular region. In the cerebellum, the Purkinje cells form scattered heterotopias throughout the cortex and in the granule cell layer. Laminar discontinuities involving the olivary nucleus are noted, which also are unique to Zellweger’s syndrome [Evrard et al., 1978; Volpe and Adams, 1972]. Studies of PEX gene knockout mice have demonstrated similar neuropathologic changes [Baes et al., 1997; Faust, 2003; Faust et al., 2001; Gressens et al., 2000; Janssen et al., 2000, 2003]. N-methyl-d-aspartate (NMDA)-mediated abnormalities may contribute to the clinical findings seen in the most severely affected persons [Gressens et al., 2000].
Multiple other abnormalities have been reported. The eyes demonstrate loss of retinal ganglion cells and gliosis of the optic nerve [Cohen et al., 1983]. Retinal pigmentary degenerative changes are associated with absent electroretinograms [Hittner et al., 1981]. Hepatomegaly (present in 78 percent of patients) with periportal fibrosis may result in significant cholestasis (in 59 percent) and jaundice, micronodular cirrhosis (in 37 percent), and hypoprothrombinemia [Heymans, 1984]. Excessive iron deposition has been noted, which diminishes with age [Gilchrist et al., 1976]. Renal cortical cysts of varied sizes are present in 97 percent of patients studied pathologically but may be missed by ultrasound analyses [Bernstein et al., 1974; Heymans, 1984]. The adrenal gland demonstrates changes similar to those in X-linked adrenoleukodystrophy, with cytoplasmic lamellar inclusions consisting of cholesterol esterified with very long chain fatty acids [Goldfischer et al., 1983]. Skeletal abnormalities include clubfoot, thumb rotation, and stippled chondral calcification of the patella and acetabulum in 50 percent of patients [Heymans, 1984].
Neonatal adrenoleukodystrophy and infantile refsum’s disease
The neonatal form of adrenoleukodystrophy was described by Ulrich et al. [1978] in a male infant with neonatal seizures and severe developmental delay. Postmortem examination at 18 months revealed adrenal atrophy and cytoplasmic inclusions typically seen in X-linked adrenoleukodystrophy; therefore, the disease was considered to be a connatal form of adrenoleukodystrophy. The multiple peroxisomal defects were first recognized in 1982 by Mobley and co-workers and substantiated later by others [Kelley et al., 1986; Partin and McAdams, 1983; Wanders et al., 1987]. Hyperpipecolicacidemia was described in 1968 by Gatfield and associates in a male infant with neurodegenerative disease and an enlarged liver, who died at 27 months of age. A marked excess of pipecolic acid was noted in the tissues and body fluids. In 1975, Danks and colleagues demonstrated excess pipecolic acid in Zellweger’s syndrome, and in 1988, Wanders and associates described a generalized peroxisomal dysfunction in that disorder, including the original case of Gatfield and associates [Wanders et al., 1988c]. The term infantile phytanic acid storage disease, a possible variant of Refsum’s disease, was used by Scotto et al. [1982] to describe the disorder in three unrelated males aged 3–6 years with hepatomegaly, dysmorphism, mental retardation, sensorineural hearing loss, and retinitis pigmentosa. The investigators noted that the liver biopsy specimen contained inclusions similar to plant chloroplasts that included phytol, which led to the measurement and recognition of increased phytanic acid levels, as in classic Refsum’s disease. The generalized peroxisomal dysfunction was documented by Poulos et al. [1984, 1986], and later by Poll-The et al. [1986a, b] and Wanders et al. [1986b].
Patients with milder forms may present in the neonatal period with mild to moderate dysmorphism, hypotonia, poor feeding, and hepatomegaly with micronodular cirrhosis. Motor and cognitive development usually is delayed. Even with hypotonia, patients may be able to walk, although gait often is ataxic. Retinal pigmentary degeneration may not become evident until the age of 4–6 months and often results in visual loss in the first years of life. Electroretinograms show profound abnormalities in nearly all affected persons and do not correlate well with vision in this population. Sensorineural hearing loss is associated with limited language development [Aubourg et al., 1986; Boltshauser et al., 1982; Budden et al., 1986; Poll-The et al., 1987; Scotto et al., 1982; Torvik et al., 1988; Wanders et al., 1990a; Weleber et al., 1984]. Adrenal dysfunction may develop with age [Govaerts et al., 1989; Poll-The et al., 1987]. Liver dysfunction often is present and detectable by persistent elevation of liver enzymes. A bleeding diathesis that responds to vitamin K also may develop, and in several children, esophageal varices were observed, consistent with portal hypertension. Several investigators have reported postmortem studies in patients aged 8 months to 12 years [Challa et al., 1983; Chow et al., 1992; Powers and Moser, 1998; Torvik et al., 1988]. The liver demonstrated micronodular cirrhosis, and the adrenal glands were hypoplastic. Lipid-laden macrophages were present in the liver, lymph nodes, and certain areas of the cerebral white matter. The only cerebral malformation was a variable hypoplasia of the cerebellar granular layer and a migrational defect of the Purkinje cells. Retinal abnormalities include a loss of ganglion cells, gliosis of the nerve fiber layer, and bileaflet inclusions in the pigment epithelium and macrophages [Cohen et al., 1983].
Optic atrophy seen in patients with neonatal adrenoleukodystrophy/infantile Refsum’s disease has been confused with Leber’s optic atrophy [Ek et al., 1986], and the association of retinal pigmentary degeneration and sensorineural hearing loss has led to misdiagnosis as Usher’s syndrome [Kelley et al., 1986; Noetzel et al., 1983].
Life span is variable, and patients have survived to adulthood [Kelley et al., 1986; Poll-The et al., 1987]. Several older patients have now been described. All identified patients have been visually impaired, with sensorineural hearing loss.
After analyzing 40 cases, Theil et al. [1992] subdivided the clinical characteristics into major and minor features. They suggest that the combined presence of three major features (present in more than 75 percent of patients) and one or more minor features (present in 50–75 percent) warrants biochemical investigation for peroxisomal disease. Other investigators have pointed out that, in view of the lack of specificity and ease of testing, strict screening criteria are not warranted, and the possibility of neonatal adrenoleukodystrophy and infantile Refsum’s disease should be considered in more children with previously undiagnosed developmental delay [Moser and Raymond, 1998].
All of the disorders in this group are inherited in an autosomal-recessive fashion, so it is important for genetic counseling that an accurate diagnosis be arrived at expeditiously. Carrier detection is possible only by DNA analysis if the molecular defect has been identified in the index case [Shimozawa et al., 1992]. Carrier detection is not possible by biochemical determination in blood or fibroblasts.
Laboratory Diagnosis
The diagnostic abnormalities present in patients with Zellweger spectrum disorders are listed in Table 38-2. A major diagnostic biochemical abnormality is the increased amount of very long chain fatty acids, which are fatty acids with carbon chains of more than 22. They also are elevated in single-enzyme defects of peroxisomal β-oxidation, so their presence is nonspecific, and other studies are required for this diagnosis. Very long chain fatty acid accumulation here is due to the reduction of peroxisomal β-oxidation, which is greater in Zellweger’s syndrome than in milder forms. A virtual absence of peroxisomal acyl-CoA oxidase and bifunctional enzyme has been demonstrated in the liver of patients with Zellweger’s syndrome [Suzuki et al., 1986; Tager et al., 1985]. It should be noted, however, that very long chain fatty acids are not elevated in rhizomelic chondrodysplasia punctata and other peroxisomal disorders (e.g., acatalasemia) that do not involve lipid oxidation, and normal levels do not exclude all peroxisomal disorders.
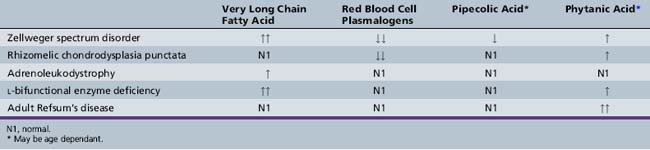
Because the first two steps of plasmalogen synthesis are peroxisomal functions, a reduction of plasmalogen levels is noted in patients with peroxisome biogenesis defects. Deficient levels of dihydroxyacetone phosphate acyltransferase have been used for diagnostic purposes in skin fibroblasts, amniocytes, chorionic villus samples, red blood cells, leukocytes, and platelets [Besley and Broadhead, 1987; Datta et al., 1984; Schutgens et al., 1985; Wanders et al., 1985, 1986a]. Webber et al. [1987] have demonstrated that the Km values and other properties of the residual enzyme are normal, suggesting that the enzyme is not defective but is abnormally labile in the cytosol because of failure of transport into the peroxisome. Plasmalogens are reduced to 5 percent of control values in Zellweger’s syndrome [Heymans et al., 1984; Wanders et al., 1986a].
An age-related increase in the levels of phytanic acid occurs in all of the peroxisome biogenesis disorders, including rhizomelic chondrodysplasia punctata, although not to the extent seen in classic adult Refsum’s disease. The accumulation of phytanic acid was demonstrated first in infantile Refsum’s disease [Scotto et al., 1982] and later in Zellweger’s syndrome [Poulos et al., 1984]. Poulos et al. [1988] demonstrated that pristanic acid is elevated in disorders of peroxisome biogenesis. This elevation was later confirmed to be due to an impaired capacity to oxidize pristanic acid [Singh et al., 1990]. These findings are unlike those in classic Refsum’s disease, in which the defect involves the conversion of phytanic acid to pristanic acid. Thus, oxidation of pristanic acid is a peroxisomal function that is disrupted in disorders of peroxisome biogenesis [Watkins et al., 1990].
Normally, bile acid intermediates, such as trihydroxycholestanoic acid and dihydroxycholestanoic acid, are absent or present in low concentrations. In Zellweger spectrum disorders, however, they constitute 30–50 percent of total plasma bile acids [Clayton et al., 1987; Eyssen et al., 1985; Gustafsson et al., 1983; Hanson et al., 1979; Mathis et al., 1980; Monnens et al., 1980; Poulos and Whiting, 1985]. Bile acid intermediates also may be abnormal in single-enzyme defects, such as peroxisomal bifunctional enzyme deficiency.
The impaired activity of l-pipecolic acid oxidase results in increased accumulation of pipecolic acid in plasma and increased excretion of pipecolic acid in urine of patients with Zellweger spectrum disorders [Kelley and Moser, 1984; Mihalik et al., 1989; Wanders et al., 1988a]. Pipecolic acid levels in blood and urine are age-dependent, but an excess can be demonstrated in all patients, irrespective of age [Dancis and Hutzler, 1986]. Medium- and long-chain dicarboxylic acids accumulate and are excreted in urine, suggestive of a partial block in the degradation of long-chain dicarboxylic acids [Bjorkheim et al., 1984; Rocchiccioli et al., 1986].
In Zellweger’s syndrome, the number of catalase-containing particles in liver or kidney biopsy specimens is reduced [Goldfischer et al., 1973; Small et al., 1988], although today biopsy rarely is needed for diagnosis. A reduction in the number of peroxisomes also occurs in neonatal adrenoleukodystrophy and infantile Refsum’s disease but, as expected, is less severe than in Zellweger’s syndrome. The catalase is cytosolic rather than particulate in catalase-containing particles. Although, in the past, peroxisomes were considered absent, Santos et al. [1988] demonstrated membranous structures called peroxisomal ghosts that contain peroxisomal membrane proteins. This finding has been substantiated by other investigators, who demonstrated the presence of major membrane proteins in skin fibroblasts from patients with Zellweger’s syndrome, some of which were quite abundant [Gaertner et al., 1991; Suzuki et al., 1989]. These ghosts lack the matrix enzyme catalase and some or all of the matrix proteins. Schram et al. [1986] have reported that peroxisomal β-oxidation enzymes, which normally are located in the peroxisome matrix, are formed at a normal rate but are rapidly degraded in the cytosol. The mitochondrial abnormalities observed are considered secondary [Trijbels et al., 1983].
Prenatal Diagnosis
Cultured amniocytes or chorionic villus cells can be used to diagnose all of the disorders of peroxisome biogenesis. A variety of biochemical strategies have been used and focus on the type and degree of biochemical abnormality. Two independent techniques, the measurement of very long chain fatty acid β-oxidation [Moser et al., 1984a; Solish et al., 1985] and plasmalogen synthesis [Roscher et al., 1985; Schutgens et al., 1985], have demonstrated accurate diagnosis when performed by laboratories experienced in these methodologies. Molecular genetic techniques may be used in families in which the mutation has been identified and pre-implantation genetic diagnosis has been performed [Al-Sayed et al., 2007].
Therapy
Attempts to normalize some of the biochemical abnormalities include oral ether lipid therapy [Holmes et al., 1987; Poulos et al., 1990; Wilson et al., 1986] and dietary restriction of very long chain fatty acids [Van Duyn et al., 1984] and phytanic acid [Greenberg et al., 1987; Steinberg, 1989]. These approaches may normalize plasma levels of very long chain fatty acids, phytanic acid, and red blood cell plasmalogens. The clinical effectiveness of these interventions, however, has yet to be demonstrated because of the phenotypic variability of these disorders. Ursodeoxycholic acid can reduce levels of bile acid intermediates and has the potential to prevent liver damage [Colombo et al., 1990]. Dietary therapy with docosahexaenoic acid replaces low levels in plasma and red blood cells of patients with Zellweger’s syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease [Martinez et al., 1993, 1994]; however, it is ineffective in improving clinical function [Mahmood et al., 2010]. Peroxisome proliferators, such as clofibrate, do not induce the formation of catalase-containing peroxisomes or improve clinical status [Lazarow et al., 1985].
Rhizomelic Chondrodysplasia Punctata
Rhizomelic chondrodysplasia punctata is characterized by severe shortening of limbs, mental retardation, and early death [Spranger et al., 1971]. A striking reduction in plasmalogen synthesis exceeding that seen in Zellweger’s syndrome led to the recognition of a peroxisomal defect in this disorder [Heymans et al., 1985, 1986]. Subsequent studies have revealed the following triad of biochemical defects:
The levels of very long chain fatty acids are normal, suggesting that the immature thiolase is catalytically active. Despite the presence of catalase in the particulate fraction, peroxisomal structure may not always be normal. Heymans et al. [1986] described two patients in whom peroxisomes were absent in some hepatocytes, but large or irregularly shaped in others.
The clinical features (Figure 38-4A) include disproportionate shortening of the proximal portions of the extremities, short stature, microcephaly, dysmorphic facial appearance, cataracts, ichthyosis, and severe mental retardation. Radiologic highlights include shortening of the proximal limbs, metaphyseal cupping, and disturbed ossification with epiphyseal and extra-epiphyseal calcification (see Figure 38-4B). Lateral views of the spine reveal coronal clefts of the vertebral bodies (see Figure 38-4C). Epiphyseal stippling involves mainly the knee, hip, elbow, and shoulder, and is uncommon in the vertebral column. The clinical features resemble those seen in warfarin embryopathy and should be differentiated from those of the milder autosomal-dominant form (Conradi–Hunermann type) and the X-linked forms of chondrodysplasia punctata, in which no biochemical abnormalities of peroxisomal function are detectable [Schutgens et al., 1988].
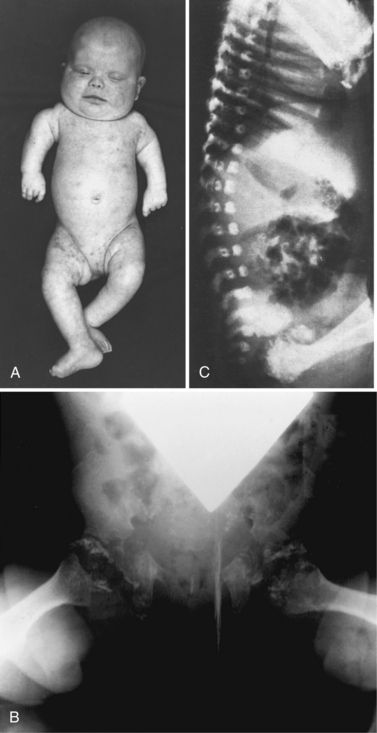
Fig. 38-4 Rhizomelic chondrodysplasia punctata
(A and B, Courtesy of Dr. B McGillivary, Vancouver, British Columbia. C, Courtesy of Dr. J Dorst, Johns Hopkins Hospital, Baltimore, Maryland.)

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


