26 Polyneuropathy
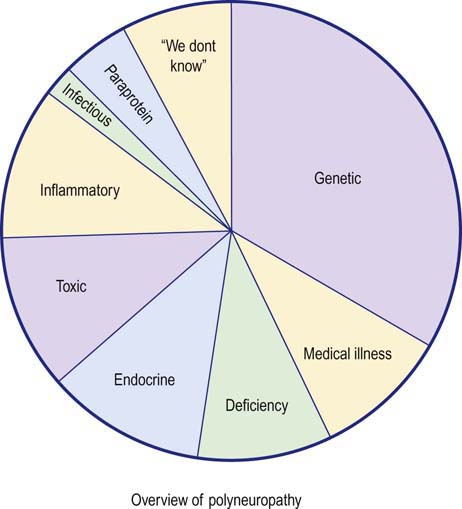
Nerve conduction studies and electromyography (EMG) play key roles in the evaluation of patients with suspected polyneuropathy. Although polyneuropathy has hundreds of potential causes, they can be grouped into several large categories (Figure 26–1). The first step in the evaluation of a patient with polyneuropathy is to reduce the differential diagnosis to a smaller, more manageable number of possibilities. This usually can be accomplished by acquiring several critical pieces of information from the history, physical examination, and electrophysiologic studies. Electrophysiologic studies can be used (1) to confirm the presence of a polyneuropathy, (2) to assess its severity and pattern, (3) to determine whether motor, sensory, or a combination of fibers are involved, and, most importantly, (4) to assess whether the underlying pathophysiology is axonal loss or demyelination. In cases in which a demyelinating polyneuropathy is found, further differentiation between an acquired and inherited condition can often be made. The information obtained from electrophysiologic testing, in conjunction with key pieces of clinical information, usually allows the differential diagnosis to be narrowed considerably so that further laboratory testing can be more appropriately applied and a final diagnosis reached.
Clinical
Key Question No. 1: What is the Temporal Course and Progression of the Polyneuropathy (Acute, Subacute, Chronic; Progressive, Stepwise, Relapsing/Remitting)?
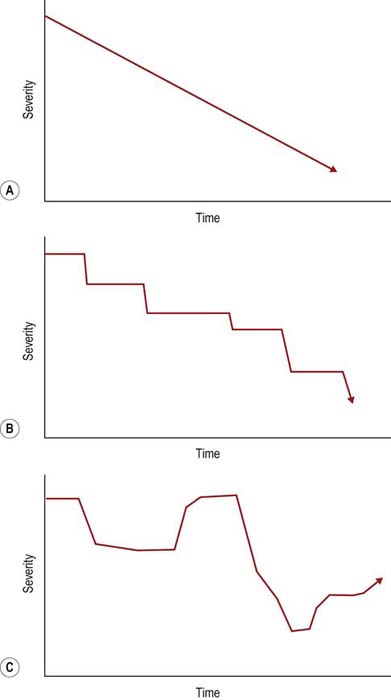
The temporal course and progression can be obtained by the history alone and often confirmed by electrophysiologic studies. Most polyneuropathies are chronic, and their onset cannot be easily determined. Acute polyneuropathies are notably less common (Box 26–1). Among them, Guillain–Barré syndrome (and its most common variant, acute inflammatory demyelinating polyneuropathy [AIDP]) is the most distinctive, with an onset over a few days or a few weeks at most. Similarly, most polyneuropathies are slowly progressive (Figure 26–2). Polyneuropathies that progress in a stepwise fashion are infrequent and are often associated with a mononeuropathy multiplex pattern (discussed later). Likewise, the history of a relapsing/remitting course is distinctly unusual and suggests either an intermittent exposure/intoxication or a variant of chronic inflammatory demyelinating polyneuropathy (CIDP).
Key Question No. 2: Which Fiber Types are Involved (Motor, Large Sensory, Small Sensory, Autonomic)?
When nerve is diseased, it can react in a limited number of ways. Thus, many peripheral nerve disorders present with similar symptoms despite different etiologies. Symptoms and signs of nerve dysfunction result either from lack of function (negative symptoms and signs) or from abnormal function or overfunctioning (positive symptoms and signs). For example, anyone who has “fallen asleep” on his or her arm can remember the initial numbness or lack of feeling (negative symptoms), followed by intense pins-and-needles paresthesias (positive symptoms) as circulation is restored. Characteristic positive or negative sensory symptoms and signs caused by diseased nerves help one recognize which fiber types are involved (Table 26–1).
Table 26–1 Negative and Positive Symptoms and Signs of Peripheral Nerve Disease
| Negative | Positive | |
|---|---|---|
| Motor | Weakness | Fasciculations |
| Fatigue | Cramps | |
| Hyporeflexia or areflexia | Myokymia | |
| Hypotonia | Restless legs | |
| Orthopedic deformities | “Tightness” | |
| (e.g., pes cavus, hammer toes) | ||
| Sensory | ||
| Large fiber | Decreased vibration sensation | “Tingling” |
| Decreased joint position sensation | “Pins and needles” | |
| Hyporeflexia or areflexia | ||
| Ataxia | ||
| Hypotonia | ||
| Small fiber | Decreased pain sensation | “Burning” |
| Decreased temperature sensation | “Jabbing” | |
| “Shooting” | ||
| Autonomic | Hypotension | Hypertension |
| Arrhythmia | Arrhythmia | |
| Decreased sweating | Increased sweating | |
| Impotence | ||
| Urinary retention | ||
Large and small fibers are affected in most polyneuropathies. Only a few polyneuropathies preferentially affect small fibers (Box 26–2). Manifestations include autonomic dysfunction and a distal sensory deficit, particularly for pinprick, often associated with painful, burning dysesthesias. It is essential to appreciate that routine nerve conduction studies assess only large myelinated fibers. A patient who has a pure small-fiber polyneuropathy, with complete sparing of the large fibers, may have completely normal electrophysiologic studies. Conversely, large-fiber polyneuropathies always show abnormalities on electrophysiologic testing. Predominantly large-fiber polyneuropathies result in clinical sensory deficits (particularly for vibration and touch), weakness, and loss of tendon reflexes, with little or no autonomic and pain/temperature sensation loss.
Key Question No. 3: What is the Pattern of the Polyneuropathy (Distal Dying Back [Distal-To-Proximal Gradient], Short Nerves, Multiple Nerves; Symmetry, Asymmetry)?
The overall pattern of the polyneuropathy is determined largely based on the clinical examination and is supplemented and confirmed by electrophysiologic studies. In most polyneuropathies, there is a distal-to-proximal gradient of symptoms and signs. Distal symptoms and findings occur in most polyneuropathies, in part indicating the frequency with which axonal loss is the underlying pathologic process. Most axonal polyneuropathies exhibit a distal-to-proximal, dying back pattern, reflecting that the chance of damage to a nerve is length dependent (Figure 26–3). Thus, the longest nerves are affected first, resulting in a stocking-glove distribution of symptoms. Patients initially develop numbness or weakness of the toes and feet, which then slowly progresses up the leg. When the process reaches the upper calf, the fingertips become involved as well, because the distance from the lumbosacral spinal cord to the upper calf is the same as that from the cervical spinal cord to the fingertips. Only rarely will polyneuropathies preferentially affect the shorter, more proximal nerves before the distal ones (e.g., in porphyria, proximal diabetic neuropathy, and some cases of inflammatory demyelinating polyneuropathy).
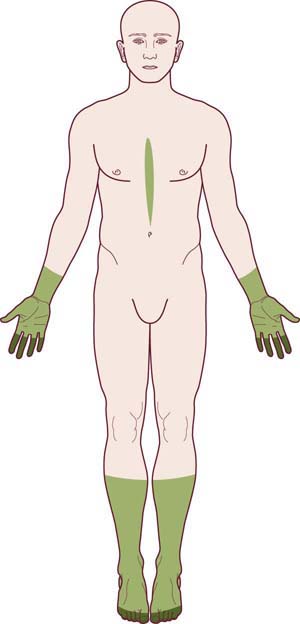
FIGURE 26–3 Stocking-glove pattern of polyneuropathy.
(Reprinted with permission from Schaumburg, H.H., Spencer, P.S., Thomas, P.K., 1983. Disorders of peripheral nerves. FA Davis, Philadelphia.)
The pattern of a mononeuropathy multiplex is one of the most important patterns to recognize and differentiate from the length-dependent, dying-back, axonal polyneuropathy. The clinical presentation is distinctive: there is an asymmetric, stepwise progression of individual cranial and/or peripheral neuropathies (Figure 26–4). Over time, a confluent pattern may develop, which may be difficult to distinguish from a generalized polyneuropathy. In most cases, the individual neuropathies are of named nerves (i.e., median, ulnar, peroneal, etc.) as opposed to small nerve twigs. Mononeuropathy multiplex has a limited differential diagnosis (Box 26–3) and most often occurs in the setting of vasculitis and vasculitic neuropathy. As each subsequent nerve is infarcted, pain develops (often severe), followed hours or days later by weakness and numbness in the nerve’s distribution. Although other organ systems are often involved, the initial clinical presentation of systemic vasculitis may involve only the peripheral nervous system. Indeed, there are now well-recognized cases in which vasculitis remains confined to the peripheral nervous system.
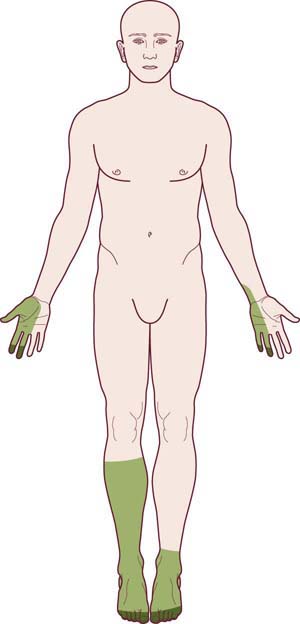
FIGURE 26–4 Mononeuropathy multiplex pattern of polyneuropathy.
(Adapted and reprinted with permission from Schaumburg, H.H., Spencer, P.S., Thomas, P.K., 1983. Disorders of peripheral nerves. FA Davis. Philadelphia.)
Box 26–3
Differential Diagnosis of Mononeuropathy Multiplex
Vasculitis (e.g., polyarteritis nodosa, Churg–Strauss syndrome, Wegener’s syndrome, hypersensitivity, cryoglobulinemia, systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome, chronic active hepatitis)
Inflammatory demyelinating polyneuropathy (Lewis–Sumner variant)
Multiple entrapments (hereditary and acquired)
Infection (e.g., Lyme, leprosy, human immunodeficiency virus)
Infiltration (e.g., granulomatous disease [sarcoid], neoplasm [lymphoma, leukemia])
Key Question No. 4: What is the Underlying Nerve Pathology (Axonal, Demyelinating, or Mixed)?
Pathologically, injury to nerves consists of two major processes: axonal loss or demyelination. The vast majority of polyneuropathies are primarily axonal. In demyelinating polyneuropathies, the initial injury to the nerves reflects damage to or dysfunction of the Schwann cells and the myelin sheaths. As a consequence of demyelination, conduction is impaired with marked slowing of conduction velocity or frank conduction block. In establishing the differential diagnosis of a peripheral nerve disorder, the presence of demyelination is always a key finding (see later). Demyelination may be demonstrated either by nerve biopsy and pathologic examination or, more easily, by electrophysiologic testing. When nerve conduction studies demonstrate a polyneuropathy to be predominantly demyelinating, the differential diagnosis is readily narrowed to a small group of disorders (Box 26–4).
Box 26–4
Demyelinating Polyneuropathies
Hereditary
Charcot–Marie–Tooth, Type I (CMT1)
Charcot–Marie–Tooth, Type IV (CMT4)
Charcot–Marie–Tooth, X-linked (CMTX)
Hereditary neuropathy with liability to pressure palsy (HNPP)
Adrenoleukodystrophy/adrenomyeloneuropathy
Cerebrotendinous xanthomatosis
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
Acquired
Acute inflammatory demyelinating polyradiculoneuropathy (AIDP, the most common variant of Guillain–Barré syndrome)
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP)
Multifocal motor neuropathy with conduction block (±GM1 antibodies)
Toxic (i.e., amiodarone, perhexiline, arsenic, glue sniffing, buckthorn shrub poisoning)
* Dejerine–Sottas disease is a historical term used to denote a severe demyelinating neuropathy in children. The classic phenotype described a hypotonic infant with areflexia and hypertrophic nerves; on nerve conduction studies, conduction velocities were extremely slow, typically around 6 m/s. Formerly considered a distinct entity with autosomal recessive inheritance, genetic analysis has demonstrated that Dejerine–Sottas is a syndrome with either recessive inheritance or autosomal dominant inheritance with de novo mutations. The recessive forms are now incorporated into the CMT4 group. The de novo autosomal dominant forms have mutations on the same genes implicated for CMT1 (P0, PMP22, and EGR2), but with the genetic defect resulting in a much more severe demyelinating neuropathy.
Key Question No. 5: Is there a Family History of Polyneuropathy?
Inherited polyneuropathies may affect certain individuals so minimally or may progress so slowly over an individual’s lifetime that the person never seeks medical attention. Therefore, it often is beneficial to examine family members, both clinically and with nerve conduction studies and EMG, to help determine whether the underlying etiology of the patient’s polyneuropathy is genetic. Several clinical clues, however, suggest the possibility of an inherited polyneuropathy (Figure 26–5):
• Foot deformity (pes cavus, hammer toes, high arches)
• History of a long-standing polyneuropathy (many years and often decades)
• History of very slow progression
• Few positive sensory symptoms
• Family history of “polio,” “rheumatism,” “arthritis,” or other disorders that actually might have been inherited polyneuropathy
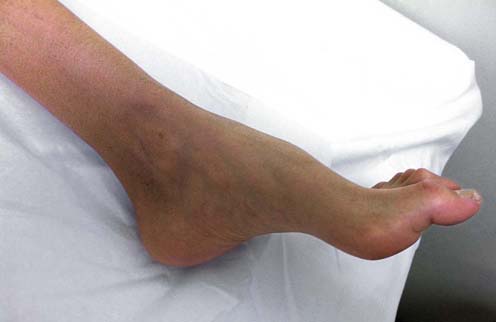
Key Question No. 6: Is there a History of Medical Illness or are there Signs Suggesting A Medical Illness Associated with Polyneuropathy?
Axonal Polyneuropathy
Electrophysiology
Special Situations in Axonal Polyneuropathy: The Use of the Sural/Radial Amplitude Ratio in Mild Polyneuropathy
1. Younger individuals have much higher baseline sural amplitudes than older individuals. Thus, if a young patient had a sural amplitude of 30 µV, then developed an axonal polyneuropathy and the sural amplitude decreased to 15 µV, this value would still be considered normal in most EMG labs.
2. Older individuals may have low or difficult to obtain sural sensory responses at baseline. Thus, in an 80-year-old patient with numbness of the feet and a sural amplitude of 3 µV, it is difficult to know whether this value indicates a neuropathy or is simply consistent with age.
3. In obese individuals, the additional adipose tissue between the skin and the underlying nerve may result in an attenuation of the sensory nerve amplitude. Thus, in an obese patient with a sural amplitude of 5 µV, it may be difficulty to know if this value indicates a neuropathy or simply denotes a reduced amplitude from technical issues related to increased intervening adipose tissue in the lower leg.
Borderline Cases: Differentiation Between Axonal and Demyelinative Slowing
In such cases, one useful technique is to compare conduction velocities recording a distal and a proximal muscle across the same segment of nerve. In the leg, the peroneal nerve is most useful for this study. Peroneal motor studies are performed stimulating below the fibular neck and at the lateral popliteal fossa and recording simultaneously from the extensor digitorum brevis (EDB), a distal muscle, and the tibialis anterior, a proximal muscle (Figure 26–6). Conduction velocities across this same segment of nerve are then compared.
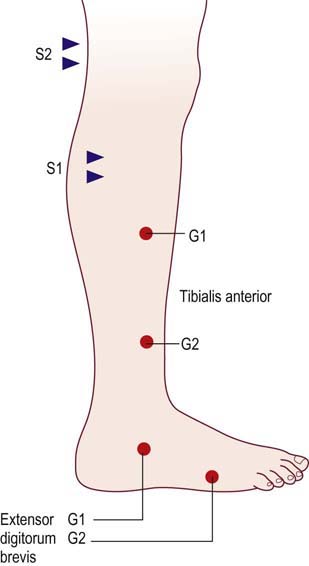
FIGURE 26–6 Recording proximal and distal muscles to differentiate axonal from demyelinative slowing.
(Reprinted with permission from Raynor, E.M., Ross, M.H., Shefner, J.M., et al., 1995. Differentiation between axonal and demyelinating neuropathies: identical segments recorded from proximal and distal muscles. Muscle Nerve 18, 402.)
In patients with demyelinating polyneuropathies, conduction velocities typically are slowed at both recording sites, with no difference between proximal and distal sites (Figure 26–7). In patients with axonal polyneuropathies, however, conduction velocities may be slowed recording the EDB but usually are normal or only mildly reduced when measured from the tibialis anterior. This distal-to-proximal gradient of conduction velocity slowing in axonal polyneuropathies may be very helpful in differentiating a primary demyelinating from axonal polyneuropathy, especially when the distal conduction velocities are near the cutoff value for demyelinative slowing.
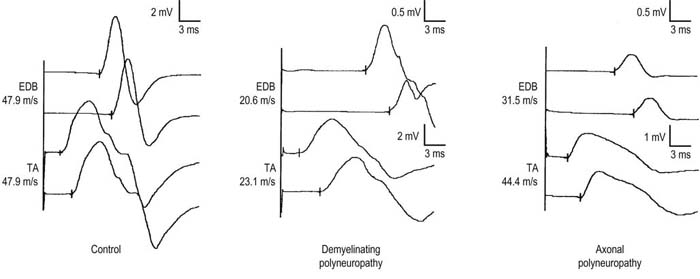
FIGURE 26–7 Proximal and distal recordings are used to differentiate demyelinative from axonal slowing.
(Reprinted with permission from Raynor, E.M., Ross, M.H., Shefner, J.M., et al., 1995. Differentiation between axonal and demyelinating neuropathies: identical segments recorded from proximal and distal muscles. Muscle Nerve 18, 402.)
Demyelinating Polyneuropathy
In addition, nerve conduction studies often can be used to distinguish between acquired and inherited demyelinating polyneuropathies. In a patient with an inherited condition, all myelin tends to be affected equally; thus, uniform slowing of conduction velocity occurs. Accordingly, nerve conduction studies usually are symmetric from side to side. In contrast, acquired conditions (e.g., Guillain–Barré syndrome, CIDP) are associated with patchy, often multifocal demyelination. As a result, asymmetry is found on nerve conduction studies (even in the face of clinical symmetry), along with evidence of conduction block and temporal dispersion. Conduction block and temporal dispersion at non-entrapment sites are key findings for differentiating acquired from inherited demyelinating polyneuropathies (Figure 26–8).
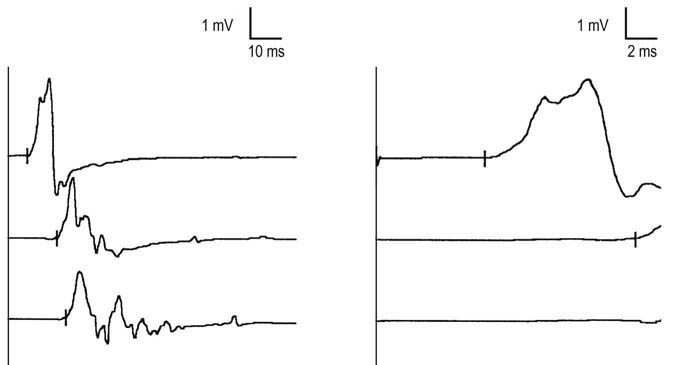
Guillain–Barré Syndrome (GBS)
Electrophysiology
To demonstrate segmental demyelination on motor nerve conduction studies, a combination of conduction block or temporal dispersion, or marked slowing of distal latencies, conduction velocities, or late responses must be seen. For acute polyneuropathies, the electrophysiologic criteria for segmental demyelination often are liberalized (Box 26–5).
Box 26–5
Electrophysiologic Criteria for Acute Demyelinating Polyneuropathy
Demonstrate at least three of the following in motor nerves:
1. Prolonged DLs (two or more nerves, not at entrapment sites)
2. CV slowing (two or more nerves, not across entrapment sites)
3. Prolonged late responses: F response and H reflexes (one or more nerves)
(Note: If distal CMAP amplitude is very low, absent F waves may not be abnormal.)
4. Conduction block/temporal dispersion (one or more nerves)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


