Schematic diagram of the investigation of thunderclap headache. CT, computed tomography; MRI, magnetic resonance imaging; MRA, magnetic resonance angiography; MRV, magnetic resonance venography; TCD, transcranial Doppler; DSA, digital subtraction angiography; SAH, subarachnoid hemorrhage; CVT, cerebral venous thrombosis; IIH, idiopathic intracranial hypertension; RCVS, reversible cerebral vasoconstriction syndrome; PRES, posterior reversible encephalopathy syndrome; SIH, spontaneous intracranial hypotension.
The pathological mechanisms involved in the production of thunderclap headache include direct arterial vessel damage by rupture, inflammation or rapid change in vessel caliber, processes that stimulate periadventitial nociceptive fibers [4]. This can happen in SAH, arterial dissection, cerebral vasoconstriction, sentinel headache, and acute hypertensive crisis. Similar pain may be reproduced during cerebral angiography and endovascular procedures, in response to specific maneuvers that increase intra-arterial pressure and/or to the contact of substances (contrast dye, glue, coils, balloons) with the vessel wall. Pain location elicited in these procedures usually reflects the anatomical distribution of nociceptive arterial nerve fibers of the manipulated vessel [4], although such topographic correlation is not as clear in vascular disease. Sudden, intense sharp head pain may also be a consequence of increased intracranial pressure or meningeal stimulation, as occurs in cerebral venous thrombosis (CVT), ischemic or hemorrhagic stroke, pituitary apoplexy, meningitis, colloid cyst of the third ventricle, hydrocephalus, or subdural hematoma. Surprisingly, spontaneous intracranial hypotension can also present as TCH [5].
There are no clinical characteristics of the headache itself that allow differentiation between primary and secondary TCH [3]. A thorough history will help the clinician in the identification of the sudden onset and/ or excruciating “worst of their life” intensity characteristics of the headaches. Any of these features is enough to warrant full etiological investigation. Accompanying symptoms that have been found to increase the likelihood of a secondary cause include nausea, impaired consciousness, seizures, and diplopia [3,6]. Vomiting, transient loss of consciousness, and focal neurologic symptoms are slightly more frequent in symptomatic (secondary) TCH [3]. Female sex, neck stiffness, and abnormalities in neurological examination are also more likely in symptomatic TCH [3,6].
Ancillary evaluation of TCH may be directed to the most probable underlying cause, given the patient’s history. This may require CT scan, LP, MRI, angiographic arterial and/or venous intracranial imaging (by CT or MRI), transcranial Doppler (TCD) or even digital subtraction angiography (DSA).
This chapter will review the most relevant cerebrovascular etiologies of TCH focusing on the worst diagnostic challenge of TCH – the approach to patients with isolated TCH, i.e., TCH with normal neurological examination.
Case 1. Focal neurological signs 3 weeks after non-remitting TCH
Case description
A 38-year-old hairdresser presented to the emergency room (ER) with an intense non-remitting headache that awakened her from sleep with immediate vomiting, five nights before. The bilateral pain had occipital location, radiated to the neck, always accompanied by nausea and frequent vomiting, photophobia, phonophobia. Headache worsened with head and neck movement and physical effort. She had a previous history of migraine without aura, cigarette smoking (23 pack-years), and oral contraceptive use (desogestrel 0.15 mg plus ethinylestradiol 0.02 mg). Her migraines had started at the age of 16, current attack frequency was four episodes monthly, lasting 2 to 3 days. Her attacks were severe and each month she had at least one to two days of work absenteeism on account of her migraine. She had never been on migraine preventive treatment and treated her attacks with ibuprofen and domperidone. She noted some differences of this headache to her usual migraine: it was stronger than usual, bilateral and posterior in location, resistant to usual analgesics and lasted longer. Her vital signs and neurological examination were normal. A CT scan performed at the ER was also normal. She was discharged after partial improvement of her pain with intravenous analgesics and metoclopramide. She was prescribed amitriptyline 25 mg daily, naproxen 500 mg with metoclopramide three times daily for 5 days, and zolmitriptan 5 mg, as rescue medication.
Over the next 2 weeks she noticed progressive improvement of the intensity of her headaches. Vomiting stopped but the pain was still disrupting enough to prevent her from returning to work. She was using zolmitriptan once or twice daily. Around 20 days after the beginning of her headache, the pain suddenly became more intense, without nausea and vomiting, and a couple of hours later she noticed weakness of her left arm and was brought to the ER. She was somnolent and her speech was slurred. She was unable to walk without assistance due to left-sided hemiparesis (central facial palsy, arm strength 2/5, leg strength 3/5). Her blood pressure was 150/85 mmHg, pulse 64 with normal rhythm.
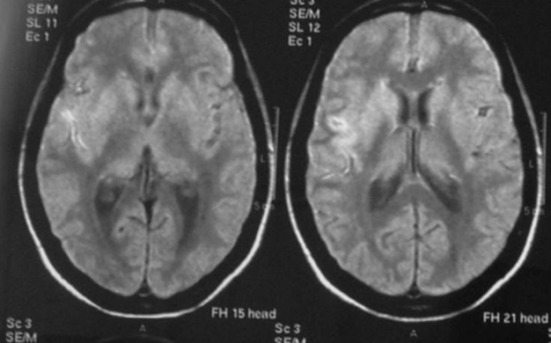
Brain CT and magnetic resonance imaging (MRI) revealed subarachnoid blood and a recent infarct of the deep territory of the right middle cerebral artery (MCA) (Figure 12.2). Magnetic resonance angiography (MRA) identified narrowing of the right internal carotid and middle cerebral arteries. TCD showed a marked increase in blood flow velocities in the right MCA (systolic velocity 370 cm/second, diastolic velocity 200 cm/second) compatible with vasospasm and mild increases in the left anterior cerebral (ACA) and basilar arteries (systolic speed 290, diastolic speed 160). Angiography confirmed marked (>50%) vessel caliber reduction in the right internal carotid and middle cerebral arteries, moderate (25–50%) spasm of the anterior cerebral arteries, and identified a single saccular aneurysm (8 × 4 mm) with wall irregularity of the right posterior communicating artery (Figure 12.3).
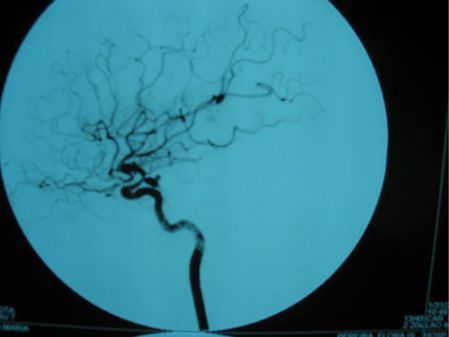
She was started on oral nimodipine, euvolemia was restored with crystalloid fluids and blood pressure was controlled, keeping her systolic values between 130 and 170 mmHg. There was rapid clinical and TCD improvement. She underwent aneurysm treatment by microsurgical clipping within 72 hours of admission. Control angiography confirmed aneurysm exclusion. She showed progressive improvement of her symptoms and was discharged with no further complications after 2 weeks, with residual left hemiparesis (modified Rankin scale 2).
Discussion
SAH is the most frequent cause of symptomatic TCH. Misdiagnosis still occurs in around 12% of cases, either due to lack of recognition of its importance (by patients and/or physicians) or to not performing a comprehensive complementary evaluation [7,8].
The most common misdiagnoses of SAH are tension-type headache and/or migraine. The risk of misdiagnosis is increased in patients who are fully conscious and without focal neurological signs (Hunt and Hess grades 1 or 2), and with previous history of migraine of hypertension. Low physician suspicion [8,9] and consequently delayed diagnosis increases the likelihood of death or disability fourfold [7].
The mode of onset of the headache is the most important feature in TCH characterization that typically occurs during or after strenuous tasks or exercise, especially in patients aged over 60 [10]. Nevertheless, the majority occur during effortless everyday routines or even during sleep, the latter often preventing both patient and doctor from identifying its sudden onset. The pain may be severe and unremitting, but also brief and self-limited, which is typically the case of the “sentinel” headache occurring in warning leaks, in 10–40% of patients [7]. Up to 20% of patients have milder headaches, isolated neck pain, or no headache at all [7]. When the headache’s onset is unclear, the high-intensity “worst of a lifetime” pain is the second clinical characteristic that mandates immediate complementary investigation. Although a normal neurological examination does not exclude any serious underlying disorder in a patient with TCH, it is important to screen for subtle neurological signs that may bring further clues to the diagnosis, such as a unilateral dilated and hyporesponsive pupil, mild nuchal rigidity, or ophthalmoscopic changes (including papilledema or peripapillar hemorrhages). These may elude the less trained general practitioner or ER doctor. The presence of focal neurological signs and changes in consciousness at presentation increase diagnostic accuracy [7,8].
The most sensitive test for the diagnosis of acute SAH is non-contrast CT scan, with sensitivity similar to MRI FLAIR, of almost 100% in the first 3 days of SAH. The sensitivity of CT decreases to around 85% by the fifth day and 30% after 2 weeks, MRI being superior to CT in this time frame. In the evaluation of TCH, a normal CT or MRI in the first 2 weeks makes it mandatory to perform a lumbar puncture to search for CSF xanthochromia. Traumatic, very early (<6 to 12 hours) or late (> 2 weeks) taps can be falsely negative [7,11]. Small (< 3 mm) aneurysms may fail detection by CT or MR angiography [7], but may be revealed by intra-arterial digital subtraction angiography (DSA). In non-perimesencephalic SAH, some patients have falsely negative findings on initial DSA, either due to technical reasons, concomitant vasospasm, thrombosis, or obliteration of the aneurysm by adjacent hematoma. Repeating DSA within 2 to 6 weeks if initial findings are inconclusive has a diagnostic yield of around 12% and is a current recommendation [11].
The inadvertent use of triptans in SAH has been described; the few cases reported documented an anti-nociceptive effect of triptans in headache due to SAH, as has happened in our patient [12,13]. The use of triptans in SAH-related headache increases both the risk of misdiagnosis and of arterial vasospasm, as triptans carry the potential of arterial vasoconstriction. Nevertheless, in the few cases described, vasospasm occurrence seemed unrelated to triptan use. In our patient, despite daily use of an oral triptan, vasospasm occurrence was delayed in relation to its most frequent timing [14].
Pitfall
This case illustrates the two major difficulties in the identification of TCH when taking the history – the typical sudden onset of the headache was missed due to headache onset during sleep and severity of pain was devalued due to a previous history of severe migraine. A normal initial neurological examination does not exclude SAH, but remember – it increases the odds for misdiagnosis.
Case 2. Recurring excruciating TCH
Case description
A 60-year-old secretary presented in the emergency room (ER) due to headache episodes that had started 10 days before. The first episode was of a progressive bilateral frontal pressure-like pain, without accompanying symptoms that persisted through the day but suddenly worsened acutely, without any precipitating factor. This excruciating pain was predominantly over the left temple, lasted around half an hour, and was accompanied by nausea and vomiting. There was no photophobia, phonophobia, worsening with Valsalva’s or with physical effort. A mild bilateral frontal pain persisted, yet she was able to function normally. Repeated and identical exacerbation episodes occurred suddenly at rest, the second time 48 hours after the first and the third 72 hours after the second, always with mild persistent pain between these episodes. She often took paracetamol (acetaminophen) and reported mild relief from baseline pain. On the ninth day, she woke up from her night sleep with the fourth sudden excruciating pain episode, accompanied by persistent vomiting, which lasted longer than one hour. She then decided to seek medical attention. Her past medical history was remarkable for mild essential hypertension controlled with bisoprolol 5 mg daily, and nephrolithiasis. She had no previous history of migraine or other primary headaches. She denied any contact with other pharmacological or toxic agents, including selective serotonin reuptake inhibitors (SSRIs), cannabis, or nasal decongestants. She had no previous complaints of fever, joint pain, skin rashes, oral ulcers, or ocular disorders except mild presbyopia in the last couple of years.
Upon admission to the ER her vital signs and neurological examination were completely normal, yet she was very uncomfortable with headache graded 10 out of 10 on a visual analog scale and was still vomiting. A CT scan and routine blood work-up were normal. An LP revealed a normal CSF without xanthochromia. Serologic (neurotropic viruses, Borrelia species) and vasculitis screen revealed a positive ANA titer of 1/320 without any other abnormalities.
Brain MRI revealed multiple T2 non-enhancing hyperintense subcortical white matter lesions, with highest expression in the right frontal area. MRA identified multiple narrowing of the distal branches of both MCAs and of the posterior circulation. TCD on the tenth day failed to disclose changes in blood flow velocities in proximal MCAs, ACAs, or basilar arteries (Figure 12.4).
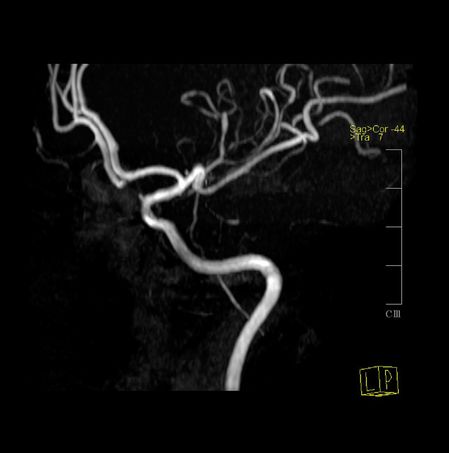
Her headache was treated with metamizole, tramadol, and metoclopramide. Oral nimodipine 60 mg every 6 hours was started, with improvement of baseline pain and no further recurrence of TCH episodes. Control MRA after 4 weeks had complete normalization of all previous changes; nimodipine was stopped after 8 weeks.
Discussion
Repeated episodes of TCH are very characteristic of reversible cerebral vasoconstriction syndrome (RCVS), an entity described since the 1970s, known to be associated with diverse conditions. RCVS causes characteristic diffuse cerebral arterial stenosis; when no cause or association is identified, common terms include idiopathic reversible cerebral segmental vasoconstriction or Call–Fleming syndrome [15,16].
One of the most important clues to the diagnosis is the epidemiological context. RCVS is more frequent in middle-aged female patients and in up to 60% of cases is precipitated by specific conditions such as the postpartum period or by vasoactive substances, including therapeutic agents (such as selective serotonin reuptake inhibitors, nasal decongestants, triptans, ergot alkaloid derivatives, intravenous immunoglobulin, red-blood-cell transfusion, or interferon alfa) but also over-the-counter or recreational substances such as ginseng and other herbal medicines, alcohol (binge drinking), cannabis, cocaine, methylenedioxymethamphetamine, amphetamines, and lysergic acid diethylamide [15].
In the case presented, the headache had two red flags for a secondary etiology: onset after the age of 50 and thunderclap-like pain. RCVS can be responsible for up to 9% of TCH cases without changes in general or neurologic examination [17], especially when repeated episodes of TCH are reported; indeed, the second important clue for the diagnosis is the temporal profile of pain. Over a background of mild to moderate persistent pain, a set of TCH exacerbations usually occur within a limited time window of 1 to 4 weeks. Seizures can occur in up to 20% of patients. The neurological examination is usually normal, unless RCVS is complicated by stroke, which produces persistent focal deficits. Meningeal signs are rare, unless concomitant convexity SAH occurs. Convexity SAH is very suggestive of RCVS in young individuals [15,16].
Differential diagnosis of recurrent TCH includes sentinel headache from SAH, CVT or other conditions that induce intracranial hypertension, primary angiitis of the central nervous system (PACNS), and several primary headache syndromes such as migraine, primary cough headache, primary headache associated with sexual activity, primary exercise headache, and primary thunderclap headache [2]. The clinical distinction between these primary or idiopathic headache syndromes and RCVS can be very challenging, as RCVS-related TCH is triggered, in 80% of patients, by sexual activity, Valsalva’s maneuver, stressful situations, physical exertion, coughing, sneezing, or other situations. Migraine-like features (nausea, vomiting, photophobia, and phonophobia) are very often present in RCVS headache and previous headache history of migraine is present in up to 40% of patients. The two important clues not to miss to the diagnosis of RCVS are the sudden onset of the attacks and the perception of the patient that the headache is different from their usual migraine attacks [15]. Mistaking RCVS headache for migraine is dangerous, as it may result in the prescription of triptans for pain control with a consequent increase in risk of brain ischemia [15].
The diagnosis of RCVS requires identification of cerebral arterial vasoconstriction by MRA, computed tomography angiography (CTA) or TCD, or catheter cerebral angiography. However, results can be normal depending on the timing of the examination, as spasm is a dynamic process that is thought to begin in small distal arteries and then progress toward medium- and large-sized vessels [18]. Vasospasm is usually only detectable by MRA after 8 to 10 days and usually peaks after 3 weeks [17]. The sensitivity of these non-invasive techniques in detecting arterial spasm is also important, as vasospasm involving distal cerebral arterial branches is not detected by TCD and sometimes not even by MRA, the gold standard being digital subtraction angiography [16]. One-fifth of patients have early MRI changes such as distal hyperintense vessels (HVs) on fluid-attenuated inversion recovery (FLAIR) imaging that are hypothesized to represent engorged distal vessels, an observation that could point to the diagnosis.
In summary, a high index of suspicion is necessary to identify RCVS in the first or second TCH episode. If all the initial ancillary evaluations are negative, follow-up is mandatory to try to document vasospasm later. At this point, it is important to decide whether or not nimodipine should be started – on one hand, symptomatic control is necessary as the headaches of RCVS are usually resistant to analgesics; on the other hand, initiating vasodilator drugs may influence future TCD results and further delay the diagnosis. It is still debatable if nimodipine provides any treatment effect except symptomatic control [15].
In the present case, it was possible to identify early distal vasoconstriction and after starting treatment with nimodipine a clear symptomatic improvement occurred, resolution was complete and uneventful, supporting the diagnosis of RCVS. RCVS has to be distinguished from PACNS, in which headache is usually accompanied by cortical symptoms (e.g.,. cognitive dysfunction), CSF analysis shows lymphocytic pleocytosis (rare in RCVS), and clinical course is progressive. The outcome of RCVS is usually benign, the course is self-limited, and most patients (70–95%) have complete resolution without residual symptoms [16]. Persistent neurologic deficits may occur with RCVS-associated stroke. Very severe cases resulting in death or dependency are rare (less than 5%), as is recurrence [16,17].
Treatment options are empirical and based on vasodilator drugs such as nimodipine, as used to treat vasospasm associated with SAH. Response to treatment is very good (up to 80%) in terms of headache resolution. The effect of nimodipine on vasospasm and stroke prevention in RCSV is less obvious. Treatment with steroids may be harmful. Treatment regimens are continued for 4 to 8 weeks, which is the time in which spontaneous resolution eventually happens. Avoidance of the offending situation in the future is recommended, as the rare recurrences described were related to re-exposures [15,16].
Pitfall
RCVS may be very difficult to diagnose because the arterial changes are dynamic (progress from small distal to medium and then proximal large-sized arteries over days to weeks), so serial evaluations are mandatory for diagnosis and monitoring.
Case 3. TCH during viral Infection
Case description
A 29-year-old woman described a one-week history of nausea, abdominal pain, diarrhea, and low-grade fever starting in her summer holidays. Three days later she had a sudden onset intense headache occurring while at rest in bed, lasting around 5 minutes and improving to a moderate tightening band-like pain, with mild photophobia and neck pain which increased upon neck movement. She visited her primary care physician (PCP), who prescribed loperamide 2 mg twice daily, paracetamol 1 g every 8 hours, and ibuprofen 600 mg as needed, for headache. Her diarrhea and abdominal pain resolved within the next week, but the headache intensity worsened and she needed to alternate paracetamol with ibuprofen every 4 hours, even at night. Pain was not worse with effort or with Valsalva. She had mild photophobia and anorexia. Pain persisted without improvement for another week so she was referred by the PCP to the ER. She had always been healthy and had no previous headache history; she was taking oral contraception (drospirenone 3 mg plus ethinylestradiol 0.03 mg).
Her neurological examination revealed prostration and mild nuchal rigidity but she had no fever nor any changes of her vital signs. Brain CT was normal. Routine blood work-up revealed a normal white blood cell (WBC) count with a relative lymphocytosis of 53%, a positive C-reactive protein (CRP) of 3.56 mg/dL (normal < 0.60), and three- to four-fold increase on liver function tests. CSF analysis revealed mild lymphocytic pleocytosis (18 cells/μL) with normal glucose and protein levels. Gram staining was negative and cultures were sterile. Opening pressure was not measured. A diagnosis of probable viral meningitis was made. Serological testing for human immunodeficiency virus, for the Herpesviridae family (herpes simplex, varicella-zoster, Epstein–Barr, and cytomegalovirus) and for Coxsackie and Echovirus was negative. The headache improved in the first 48 hours after admission. On the third day after admission, she had a sudden episode of tingling and numbness of the right hand that progressed to the arm, then neck and right side of the face over around 5 minutes; she then noticed difficulties in her speech and had some paraphasias that lasted 3 minutes. Her neurological examination after this event was completely normal, including normal funduscopic examination.
A brain MRI and magnetic resonance venography (MRV) identified a superior longitudinal sinus thrombosis (Figure 12.5) with bilateral parietal cortical lesions compatible with small venous infarcts. She was started on subcutaneous low-molecular-weight heparin (LMWH) and had complete resolution of her headache after 24 hours. She maintained oral anticoagulation for 6 months. Screening for thrombophilia was negative.
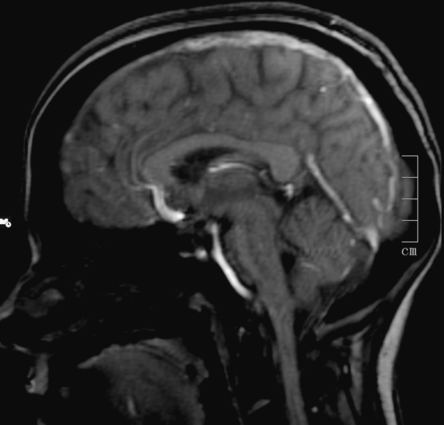
Case 3: brain magnetic resonance imaging. Sagittal T1 MRI showing thrombosis of the superior sagittal sinus.
Related posts:
 Choosing appropriate patients for anticoagulation
Choosing appropriate patients for anticoagulation
 Choosing the right patients for carotid artery procedural interventions
Choosing the right patients for carotid artery procedural interventions
 Pitfalls in the diagnosis of subarachnoid hemorrhage
Pitfalls in the diagnosis of subarachnoid hemorrhage
 When the diagnostic image is not diagnostic
When the diagnostic image is not diagnostic
 Antithrombotic management of recurrent atherothrombotic cerebrovascular disease
Antithrombotic management of recurrent atherothrombotic cerebrovascular disease
 Vascular dementia and vascular cognitive impairment
Vascular dementia and vascular cognitive impairment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


