CHAPTER 227 Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Cerebral palsy (CP) is the leading cause of developmental disability in children, with more than 500,000 individuals affected in the United States alone.1,2 CP is diagnosed in 1 in 500 live births, and this incidence continues to increase because of improvements in the care and survival of very-low-birth-weight infants.1 There have been significant advances in both medical and surgical treatment of CP spasticity in recent years. From the introduction of injectable botulinum toxin A (Botox) to the implantation of pump devices for chronic intrathecal baclofen administration, patients now have a number of pharmacologic tools to treat spasticity on a long-term basis. Permanent reductions in CP spasticity, however, can be achieved only with selective dorsal rhizotomy (SDR). Currently, a large volume of literature is available on SDR as a therapeutic modality for CP. This chapter attempts to synthesize the reported information on SDR, as well as our own experience with the procedure.
Pathogenesis of Spastic Cerebral Palsy
Spastic CP is the most common subtype of cerebral palsy. Spastic diplegia and spastic quadriplegia affect approximately 60% of patients, but spasticity may also occur with dyskinesia or ataxia, or both, in mixed subtypes of CP. Thus, in total, spasticity afflicts 80% to 90% of all patients with CP.3,4 A distinctive epidemiologic feature of spastic CP—and spastic diplegia in particular—is its correlation with premature birth and low birth weight.5–7 This relationship does not exist with other subtypes of the disease, such as athetoid or dyskinetic CP.
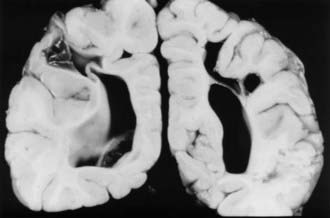
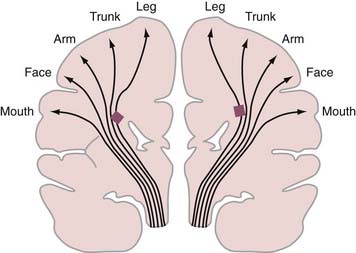
Periventricular leukomalacia (PVL) is the most common finding on brain magnetic resonance imaging (MRI) in children with spastic CP.8,9 Histologically, PVL is characterized by ischemic necrosis of the periventricular and subcortical white matter (Fig. 227-1), areas that are predisposed to hypoperfusion during hypotension.10 Reperfusion injury resulting from oxygen free radicals, cytokines, and excitatory amino acids such as glutamate probably mediate PVL.10 The high incidence of spastic diplegia in infants with PVL has led many to speculate that PVL is the neuropathologic basis for spastic CP.11 The neurological deficits in spastic CP may be explained in part by the topographic arrangements of projection fibers descending from the motor homunculus in the white matter. As seen in Figure 227-2, the leg fibers traverse the white matter closer to the ventricle than the arm fibers do. Thus, PVL is more likely to injure the leg fibers and spare the arm fibers. However, extensive leukomalacia will also injure other projection fibers and result in spastic quadriplegic CP.
Spinal cord ventral horn alpha motor neuron output is a primary determinant of muscle tone. The activity of alpha motor neurons is modulated by excitatory influences (e.g., Ia fibers from muscle spindles, descending input from the corticospinal and reticulospinal pathways) and inhibitory influences (e.g., descending GABAergic [γ-aminobutyric acid] pathways from the cerebellum and basal ganglia). Although the neural pathways involved in CP spasticity remain speculative, it is believed that spasticity results from an imbalance in these excitatory and inhibitory influences that creates hyperactive alpha and gamma neurons with exaggerated stretch reflex activity compounded by loss of inhibitory control (reviewed by Koman and colleagues12). Thus, excessive alpha motor neuron activity of the target muscles, coupled with decreased inhibition of antagonistic muscles, results in the co-contraction of agonist and antagonist muscle groups.13 Clinically, this results in a velocity-dependent, increased resistance to passive stretch.12
Deleterious Effects of Cerebral Palsy Spasticity
CP spasticity has harmful, long-term effects on growing children and thus requires optimal treatment at an early age. When untreated, inhibition of motor activity and limitation of voluntary movement result in decreased muscle and bone development, which ultimately causes deformities of the affected extremities.14 Long-lasting improvements in motor function are observed after SDR, thus supporting the argument for early intervention in the disease.15 Indeed, the spasticity-induced inhibition of motor activities does not abate if the spasticity is left untreated. Normally, longitudinal muscle growth proceeds as new myofilaments and sarcomeres are added to the ends of the muscle fibers.16 The greatest increase in sarcomere numbers occurs at an early age, with the increase taking place much more actively during development than after maturation. In parallel with the addition of sarcomeres, longitudinal muscle growth occurs rapidly during development. The increase in sarcomere number is not under neural control but is induced mainly by the amount of tension that the muscle is subjected to (i.e., repeated muscle stretch).17,18 Thus, when muscles are immobilized, there is a rapid decrease in the number of already existing sarcomeres, as well as a decrease in the number of new sarcomeres that should increase with normal development.19 Because CP spasticity in developing children limits muscle stretch in daily activities, it could cause loss of sarcomeres and inhibition of longitudinal muscle growth. In fact, the effect of spasticity on muscle growth was clearly shown in an experimental study in which longitudinal muscle growth was reduced by 45% in spastic mice as compared with control animals.20 The loss of sarcomeres accompanies a significant decrease in elasticity in the muscles21 and makes them resistant to passive stretch.22 Additionally, atrophy of type I and II muscle fibers compounds the slowed muscle growth.23 Recent work has implicated both shorter muscle (decreased sarcomere length, myocyte length, and fascicle length)24 and an increased extracellular elastic modulus25 in the clinical finding of stiffness. These pathologic events in the muscle cells cause abnormally shortened muscles, long tendons, and muscle contracture (i.e., increased resistance of the muscle to passive stretch in the absence of muscle contraction).22,26,27 The muscle contractures typically worsen as a child grows26 and produce various bone and joint deformities in the extremities.14
Treatment of Spastic Cerebral Palsy
Spasticity is commonly treated with oral medications, including the GABAB receptor agonist baclofen (Lioresal), benzodiazepines such as diazepam (Valium), the anticholinergic trihexyphenidyl, dantrolene, and tizanidine (Zanaflex).28,29 In general, oral medications are moderately effective in treating spasticity but frequently have significant adverse effects (sedation, confusion, dependence).
Intramuscular injections of alcohol and phenol for chemodenervation have been performed for many years. The introduction of intramuscular injection of Botox was a major advance in the treatment of CP spasticity, with predictable benefits and fewer adverse effects.29 Botox is injected directly into the affected muscle, and relaxation is generally seen within several days. Maximal benefit occurs approximately 4 weeks following injection, after which the effect declines and repeated injection is required in approximately 3 to 4 months.
Intrathecal Baclofen
Intrathecal administration of baclofen offers the benefit of delivering the medication directly into the central nervous system (CNS) without systemic side effects. Chronic intrathecal baclofen infusion (CIBI) with a surgically implanted pump device (SynchroMed Infusion System, Medtronic, Inc., Minneapolis, MN) has been shown to reduce spasticity and improve function,30–33 and its use has been indicated in nonambulatory or minimally ambulatory patients with spastic quadriparesis. The potential adverse effects with CIBI are significant and include infection, pump malfunction, and life-threatening withdrawal or overdose.
Stereotactic Intracranial Procedures
Stereotactic ablative procedures such as thalamotomy and dentatotomy have been used to treat CP patients with primarily unilateral dystonia and may offer some benefit in these patients.34–36 More recently, deep brain stimulation of the internal globus pallidus and thalamus have been performed in children with dystonia and tremor, respectively.37 The role of these procedures for CP spasticity has not been investigated.
Selective Dorsal Rhizotomy
SDR has been shown in several controlled trials to reduce spasticity and increase range of motion.15,38–41 In conjunction with physical therapy, SDR produces significant improvements in gross motor function and gait.15,41 SDR also decreases the rate of subsequent orthopedic surgeries.42
Indications
Indications for SDR vary among medical centers. Our current indications for SDR are summarized in Table 227-1. Primary beneficiaries of SDR are children with spastic diplegia secondary to premature birth; however, children with spastic quadriplegic CP can also benefit from SDR. In spastic hemiplegic CP, spasticity is not a predominant cause of the motor impairment, and reduction of spasticity may not greatly improve motor function (see also Table 227-2). The optimal age for children to undergo SDR is 2 to 6 years. Because of the significant deleterious effects of spasticity outlined earlier, early treatment is recommended to reduce the chance of severe orthopedic deformities of the lower extremities. SDR is not considered for children younger than 2 years because approximately 30% of children in whom CP is diagnosed at the age of 1 year later become free of symptoms.43 Adolescents and adults younger than 40 years can also enjoy good functional gains with SDR.45 The risk associated with dorsal rhizotomy in adolescents and adults is no greater than that in young children when performed through a single-level laminectomy. It is possible that reduction of spasticity will lessen the impact of aging on the physical stress caused by CP, such as abnormal stress on bones and muscles, wear and tear on joints, and increasing joint and muscle pain.
TABLE 227-1 Indications for Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
TABLE 227-2 Contraindications to Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Contraindications
Table 227-2 provides a partial list of contraindications to SDR. Such contraindications include a history of severe congenital hydrocephalus or severe neonatal CNS infection such as intrauterine encephalitis (e.g., toxoplasmosis or cytomegalovirus infection) or bacterial meningitis. Likewise, patients with severe head trauma or hypoxic encephalopathy may not be suitable candidates for SDR. Although SDR is contraindicated in patients with extensive neuronal migration disorders, those with limited schizencephaly involving the sensory motor area, a rare cause of spastic diplegia, may achieve a reduction in spasticity and functional improvements after SDR.
Patients with damage to the basal ganglia deserve special consideration because rigidity and dystonia may coexist with spasticity in this setting. Of particular concern is severe basal ganglia damage in children younger than 5 years. Dystonia may develop anytime in the first 5 years of life.46 As noted earlier, patients with mixed types of CP spasticity (i.e., those with components of dystonia or athetosis) do not enjoy the same degree of functional improvement after SDR. Thus, in the setting of basal ganglia damage it is best to wait until the age of 5, when the predominance of dystonia can more reliably be ascertained. After the age of 5, however, SDR may be considered even in the presence of demonstrable basal ganglia damage if spasticity is the primary clinical symptom. Likewise, athetosis and ataxia are also relative contraindications. Similar to dystonia, if spasticity is the predominant feature in patients with concomitant athetosis, SDR may be of benefit. In these cases, SDR reduces spasticity postoperatively, but the athetosis and dystonia remain unchanged.
Preoperative Evaluation
Motor strength can be determined by testing individual muscles. In young children, past motor milestones, speed of movement, and the ability to isolate joint movements are reliable indices of motor strength. If the motor milestone is close to normal and, for instance, the child can sit alone by the age of 2 years, the child most likely has adequate motor strength and will walk in the future. Crawling or walking in a walker and making rapid transitions between positions are also signs of relatively good motor function. As a rule, the greater the motor impairment, the slower the movement. In our experience, a neurological sign that can be an excellent predictor of gait outcome in children younger than 3 to 4 years is isolated joint movements in the lower extremities (see “Gait” in the section “Outcome”).47
Surgical Considerations and Operative Technique
Between 1987 and 1991, I (T.S.P.) performed dorsal rhizotomies on 173 patients through a laminectomy at the L2-S2 levels, as described by Peacock and colleagues.48 In 1991, however, the potential risk for late spinal deformities after the procedure led to the development of a dorsal rhizotomy procedure through an L1-2 or L1 laminectomy.49 Aside from minor technical refinements,50 the surgical technique described here has been performed between 1991 and 2008 on 1719 children and adults, with a single instance of cerebrospinal fluid (CSF) leakage and no other postoperative complications. Please refer to Tables 227-3 and 227-4.
TABLE 227-3 Subtypes of Spastic Cerebral Palsy Treated by Selective Dorsal Rhizotomy
| SPASTIC CEREBRAL PALSY SUBTYPES | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Diplegia | 1358 | 78 |
| Triplegia or quadriplegia | 356 | 21 |
| Hemiplegia | 5 | <1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
TABLE 227-4 Prerhizotomy Ambulatory Function in Patients Who Underwent Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
| AMBULATION | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Walking with a walker | 719 | 42 |
| Walking with crutches or a cane | 124 | 7 |
| Independent walking | 557 | 32 |
| Some locomotion | 180 | 11 |
| Crawling | 117 | 7 |
| No independent mobility | 22 | 1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
Operative Exposure of the Conus Medullaris
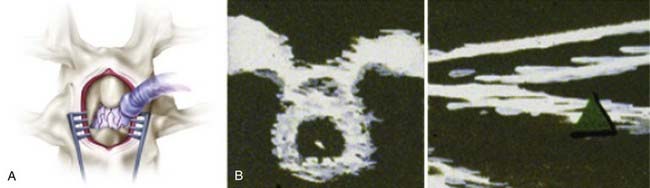
An incision is made in accordance with the localization just described. Electrocautery is used to expose the spinous process and lamina. In children, ultrasound is again used to examine the intradural structures through the exposed interlaminar space. In adults, a keyhole laminotomy may be performed to improve visualization with ultrasound. The conus is easily distinguished from the cauda equina with ultrasound (Fig. 227-3). Sagittal views show the conus as a hypodense triangle tapering caudally, whereas the cauda equina appears hyperdense. On axial views, the conus appears as a hypodense circular structure centered within the dural sac. A narrow gap between the dorsal and ventral spinal roots, lateral to the conus, can also be appreciated on the axial view. In general, axial views localize the conus more reliably than do sagittal views. A single-level laminectomy is performed to visualize 5 to 10 mm of the caudal conus and provide adequate exposure to safely separate the dorsal roots from the ventral roots later in the operation. The laminectomy may be extended to include a portion of the lamina above or below to achieve this exposure. Finally, a Midas Rex craniotome with a B5 attachment (Midas Rex Pneumatic Tools, Inc., Fort Worth, TX) is used to extend the laminectomy to the medial margin of the facet. The location of the conus is confirmed with ultrasound one final time before opening the dura. A midline durotomy is then made sharply, and the dural edges are tacked back with sutures.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


