Chapter 56 Supratentorial Tumors in the Pediatric Population
Multidisciplinary Management
Epidemiology
In the pediatric population, primary central nervous system (CNS) malignancies are the most common solid organ tumor, totaling approximately 20% of cancers in children ages 1 to 4 years and approximately 30% in children ages 5 to 9 years. In the United States, there are more than 3700 newly diagnosed cases of CNS tumors per year in children below 19 years of age. With a 66% 5-year survival rate for this age group, it is estimated that 26,000 children in the United States have a CNS tumor. The incidence rate for CNS tumors is higher in males than in females (29.9 vs. 25.1 per million) and is greatest in Caucasian individuals (29.4 per million). Based on the World Health Organization (WHO) classification system, 75% of all primary childhood CNS tumors are of neuroepithelial origin (gliomas). These include astrocytic tumors, embryonal CNS tumors, neuronal and mixed neuronal–glial tumors, and ependymomas. Ependymomas account for 15% of nonastrocytic neoplasms in the pediatric supratentorial space. Supratentorial tumors constitute 31% of pediatric CNS tumors (Table 56-1).1,2
Table 56-1 Breakdown of the Anatomic Location of Pediatric Supratentorial Tumors
| Supratentorial Brain Lesions | Patients |
|---|---|
| Temporal lobe | 7% |
| Frontal lobe | 6% |
| Multilobar | 6% |
| Ventricle | 6% |
| Parietal lobe | 4% |
| Occipital lobe | 2% |
| Total patients | 31% |
Matula C. Tumors of the pineal region. In Rengachary SS and Ellenbogen RG (eds), Principles of Neurosurgery, 2nd ed. Philadelphia, Elsevier, 2005.
Clinical Presentation
The clinical presentation is often determined by the tumor type and location, because symptoms are produced by local invasion, compression of adjacent structures, and increased intracranial pressure. More than 50% of patients have had symptoms for 6 months or longer at the time of diagnosis.3 They can be broadly categorized into generalizing and localizing symptoms. Generalizing symptoms are due to increased intracranial pressure, often a result of blockage of normal cerebrospinal fluid (CSF) flow. The most common generalizing symptoms for childhood CNS tumors include headache, nausea and vomiting, and lethargy.4 Localizing symptoms result from direct irritation, destruction, or impairment of neuronal function caused by the tumor. These include seizures, endocrinopathies, hemiparesis, cranial nerve deficits, and visual field defects5 (Table 56-2).

When considering the youngest pediatric patients, the constellation of symptoms differs because children are often unable to articulate their complaints. In this case, a thorough history with the caregiver and a physical exam are essential to uncover subtle and nonspecific signs. For children under the age of 4, the most commonly found symptoms include macrocephaly, nausea, vomiting, irritability, and lethargy.4
In the case of supratentorial tumors, symptoms are generally nonspecific and children typically have a delayed diagnosis when compared to those with infratentorial tumors. However, seizures are often seen in low-grade supratentorial lesions, either as an isolated finding (30% of patients) or with other symptoms, such as changes in personality, cognition, and development.2
Though headache is the most common manifestation of CNS tumors in children, its character is nonspecific and can vary from focal to diffuse. Thus, headaches in pediatric and adolescent patients are often attributed to benign causes, including migraines or tension headaches. However, on closer examination, these patients often present with other neurologic symptoms. Therefore, children with headaches in the setting of another neurologic deficit require further evaluation with neuroimaging. The options for imaging children include ultrasound if the fontanelle is open; x-ray, which can be of limited use in select cases; computed tomography (CT); magnetic resonance imaging (MRI); and if vascular abnormalities are suspected, angiography. The use of CT scans should be limited in the pediatric population, given the increasing evidence for radiation-induced malignancies that occur many years after image acquisition.6 If neuroimaging does confirm the presence of a mass, tissue diagnosis is often necessary to determine the appropriate treatment regimen.4
Low-Grade Glioma
Background
Pediatric low-grade gliomas, consisting of WHO grade I to II tumors, are a heterogeneous group of lesions that include pilocytic astrocytomas, oligodendrogliomas, and mixed neuroglial tumors, with pilocytic astrocytomas predominating. The overall prognosis is favorable with an indolent course and a remote risk of malignant transformation.5 While rare, there have even been case reports of spontaneous regression of low-grade astrocytomas.7,8 Low-grade astrocytomas account for 30% to 50% of all pediatric CNS tumors,9 with hemispheric (10%-15%), deep midline (10%-15%), and optic pathways (5%) being the most common locations.
The two most common pediatric low-grade gliomas are pilocytic astrocytomas (WHO grade I) and diffuse fibrillary astrocytoma (FA) (WHO grade II). Pilocytic astrocytomas typically occur in children between the ages of 5 to 19, whereas only 10% of FAs occur before the age of 20.5 Other less common pathologies include pilomyxoid astrocytoma, pleomorphic xanthoastrocytoma (PXA), ganglioglioma, subependymal giant cell astrocytoma, and oligodendroglioma. Pilomyxoid astrocytomas (WHO grade II) were first introduced in 1999; tend to occur early in life, with a median age of 18 months; and typically occur in the hypothalamus.10 Gangliogliomas and PXAs, both WHO grade II, typically occur in the temporal lobe and can present with seizures. Subependymal giant cell astrocytomas are WHO grade I lesions that occur almost exclusively in patients with tuberous sclerosis. Oligodendrogliomas (WHO grade II) are rare and account for less than 5% of brain tumors in children. When they do occur, they typically are found in the frontal lobe.5
The molecular triggers that lead to formation of low-grade gliomas are poorly understood. Few recurrent genetic changes have been described. Karyotype analysis has only revealed gain of chromosome 7 in a minority of tumors.11,12 Recently, groups have identified recurrent duplications in 7q34 in a large fraction of pilocytic astrocytomas, implicating BRAF and the RAS/RAF/MEK pathway in the pathogenesis.13,14
Pilocytic astrocytomas are the most common glial tumor in the pediatric patient and can occur anywhere in the CNS. The most common supratentorial location is the optic pathway, contributing to approximately 60% of optic pathway gliomas.5 These lesions are typically diagnosed early, with 75% of optic pathway gliomas being diagnosed before 10 years of age. More than 70% of children with optic pathway gliomas have neurofibromatosis type 1 (NF1), and many of those with NF1 develop bilateral optic nerve lesions.15
Optic Pathway Gliomas
Epidemiology
Optic nerve gliomas comprise approximately 1% of all intracranial tumors, are most commonly unilateral, and occur more frequently in females.16,17 While they can present at any age, 75% become symptomatic in the first decade of life and 90% become symptomatic before age 20.18 Rush et al. reported that in their series of 33 patients that the median age at diagnosis was 6.5 years and the mean age was 10.9 years, with a range of 2 to 46 years.19 The signs and symptoms of optic nerve gliomas typically consist of decreased visual function, proptosis, optic disc swelling, and strabismus.17 Patients can have vascular compromise of the optic apparatus from chronic compression of the central retinal vein, leading to occlusion, venous stasis retinopathy, optociliary shunt vessels, or neovascular glaucoma.17 In rare cases, acute loss of vision can occur in the setting of tumor hemorrhage. Patients typically do not present with orbital or ocular pain.17
A firm link has been established between NF1 and optic pathway gliomas. The incidence of NF1 among patients with optic nerve or chiasmal gliomas has ranged from 10% to 70% based on the series, while the incidence of optic nerve glioma in patients with NF1 varies from 8% to 31%.17,20
Diagnosis
The diagnosis of an optic nerve glioma can largely be made by either CT or MRI. The radiographic appearance varies depending on whether the person has NF1. In patients without NF1, the optic nerve is almost always enlarged in a fusiform manner with a clear-cut margin produced by the intact dural sheath. In patients with NF1, the nerve is irregular, with kinking and buckling secondary to the mass.17 On MRI, the tumor is typically hypo- to isointense on T1-weighted imaging and hyperintense on T2-weighted imaging. After intravenous administration of gadolinium-based contrast agents, the glioma may not enhance, have patchy enhancement, or demonstrate avid enhancement (Fig. 56-1).21 Additionally, MRI imaging may demonstrate abnormalities that extend beyond the optic nerve into the chiasm.17 Patients may have an enlarged optic canal ipsilateral to the side of the lesion, but this does not always indicate extension of the tumor intracranially. Arachnoid hyperplasia may be sufficient by itself to cause canal enlargement. Conversely, a normal caliber optic canal does not rule out the possibility of tumor extension beyond the orbit.17
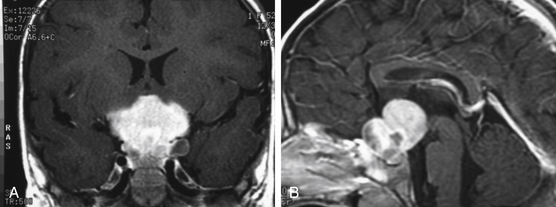
The natural history of optic nerve gliomas is usually benign, with most growing slowly in a self-limited manner or, in extremely rare cases, spontaneously regressing.8 As a result, many patients retain excellent or maintain existing visual function without treatment. Rarely, WHO grade III and IV lesions arise from the optic nerve and result in rapid visual loss, neurologic deficit, and eventual death. These high-grade lesions almost always occur in adults.22
Pathology
The gross appearance of an optic nerve glioma often causes diffuse expansion of the nerve that may extend the entire length of the nerve or affect only a portion. The expanded portion can be solid or gelatinous, with hemorrhagic and necrotic regions. In children with NF1, the tumor not only expands the nerve but often breaks through pia mater and enters the subdural space.17,23 However, as long as the glioma lies within the confines of the orbit or optic canal, the tumors are typically limited to within the optic dural sheath. If the glioma extends intracranially, it can remain intraneural or become an expansile mass that can compress adjacent structures, including the chiasm or contralateral optic nerve.17
The most common type of optic nerve tumor is the pilocytic astrocytoma, which has three predominant histologic patterns: (1) coarsely reticulated, (2) finely reticulated, and (3) coarsely fibrillated or spindle cell.21 The most common pattern is coarsely reticulated, with a biphasic pattern histologically of coarse bipolar astrocytes that are either tightly compacted around blood vessels or loosely associated around microcystic spaces. These tumors frequently have Rosenthal fibers and eosinophilic granular bodies. Finely reticulated pilocytic astrocytomas can be confused with WHO grade II diffuse low-grade astrocytomas and demonstrate an expansion of the indigenous neuroglia of the optic nerve. Within the finely reticulated tumors, a delicate reticulated syncytium of neuroglia fibers is embedded among multiple, small, round or ovoid nuclei. The coarsely fibrillated variant consists of coarse neuroglial fibrils and spindle cells arranged in bundles and is more often seen in adults.21
Management
The three major treatment modalities are surgery, chemotherapy, and radiation therapy. Surgery still plays a role in the management of optic pathway gliomas, especially in individuals without NF1 for whom MRI findings are not compelling and tissue diagnosis is required.24–26 Often in those cases, stereotactic or open biopsies can be safely performed. However, in cases where a large tumor creates mass effect or hydrocephalus, recurrent tumors refractory to chemo- or radiotherapy or cystic lesions with compression of the optic pathway can benefit from more radical surgery.25,26 Advocates of aggressive surgical resection argue that radical tumor excision allows long-term remission, given the slow-growing nature of pilocytic astrocytomas. Additionally, it could delay the initiation of radiation therapy, which has significant long-term sequelae in children. While some studies have demonstrated that aggressive resection can be safely performed if done carefully,27,28 the outcomes after surgery remain variable. For example, Sawamura et al. reported on the visual outcomes after 26 cases treated surgically with the intent to debulk as much tumor as possible.25 Of this group, 20 patients underwent radical resection with greater than 90% tumor removal, and the remaining 6 patients had partial resection. Of the 26 children, only 2 had visual improvement, 2 remained static, 10 remained blind, and 12 worsened.
Thalamic Glioma
Pediatric thalamic gliomas arise as primary gliomas or secondary gliomas from adjacent structures, including the cerebral hemispheres, caudate nuclei, brain stem, or pineal gland. The deep, central location of the thalamus makes surgical treatment of these tumors difficult without significant patient morbidity and mortality. Even though thalamic gliomas are rare and account for 1% to 1.5% of all CNS tumors, both low-grade, well-circumscribed lesions and high-grade, diffuse, infiltrating gliomas have been described.29 Although pilocytic astrocytomas classically occur in the hypothalamic/optic pathway, they can also occur as unilateral thalamic masses. Higher-grade FAs also occur in this region and are typically unilateral, with little involvement of the visual pathways. Finally, bilateral infiltrating astrocytomas, a rare variant of FAs, are lesions in bilateral thalami that have a poor outcome due to their intrinsic aggressive biology and the difficulty in attaining adequate surgical debulking of the tumor, resulting in mass effect on the thalamus.30,31 Treatment considerations concerning the necessity, feasibility, and safety of surgery in children with these lesions are varied.
Epidemiology
Thalamic gliomas are rare and account for 0.84% to 5.2% of pediatric intracranial tumors.29 This discrepancy in incidence is due to difficulty in differentiating primary thalamic tumors from secondary lesions that grow to involve thalamic structures. In a retrospective analysis of 69 children with thalamic tumors, 32 had low-grade thalamic tumors and 22 had high-grade tumors; the remaining 15 had 9 bilateral thalamic and 6 thalamopeduncular tumors. It was found that low-grade lesions had statistically improved survival rates, particularly when patients had symptom duration longer than 2 months and tumor excision greater than 90%.32
Pathology
Primary thalamic gliomas include pilocytic astrocytomas, FAs, and bilateral infiltrating astrocytomas. Pilocytic astrocytomas are low-grade gliomas, with histologic grade I. FAs are a heterogeneous group of astrocytic cells and are graded by the WHO from grade II to grade IV. They are prone to malignant progression. Bilateral infiltrating astrocytomas are a rare variant of FAs histologically that are highly infiltrating but well-differentiated grade II tumors.30 They are extremely rare, and though benign and well differentiated, patients have a poor prognosis due to thalamic nuclei involvement and inadequate surgical resection.31 It is unknown whether these bilateral tumors arise from one side of thalamic nuclei and cross the midline to the other or arise independently from tumors in the subependymal region of the third ventricle.
Diagnosis
Clinical signs and symptoms include weakness, increased intracranial pressure, motor deficits, seizures, ophthalmologic abnormalities, and in some cases, mental deterioration and personality change. It has been reported that the majority of patients have symptoms of increased intracranial pressure, headache, and vomiting.33 Though the thalamus plays an important functional role in movement, only 12% of patients have movement disorders such as tremors and dystonia. More commonly, 30% of patients present with epilepsy, despite the tumor’s deep-seated location. Another common finding in thalamic lesions includes contralateral paresis that affects one or both extremities.34 In addition, clinical presentation is correlated with tumor location. Specifically, tumors of the anterolateral thalamus are associated with sensation deficits and paresis, while those of the posteromedial thalamus are associated with early hydrocephalus.35 The presentation can also include deficits in vision such as hemianopia, a mitotic poorly reactive pupil, and ipsilateral oculomotor palsy.34
Patients typically undergo MRI prior to treatment and during follow-up. Thalamic lesions are heterogeneous and often cystic, with calcification, edema, and contrast enhancement on T1- and T2-weighted MRI32 (Fig. 56-2). The most common neuroimaging finding is hydrocephalus. Imaging is helpful in distinguishing between pilocytic astrocytomas and more infiltrative FAs. Pilocytic astrocytomas are characteristically well circumscribed, solid, and contrast enhancing. Grade II FAs are diffuse and hyperintense on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, and grade IV FAs enhance with contrast. Bithalamic infiltrating astrocytomas present as symmetrical T2 signal hyperintensity in both thalami.36
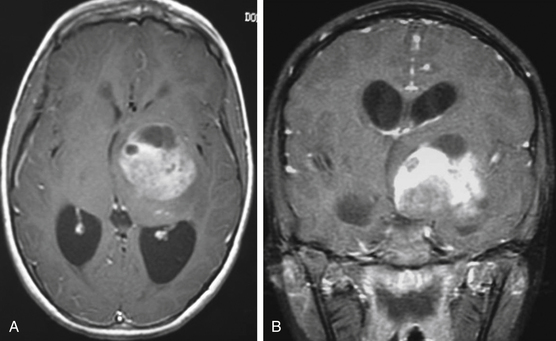
Management
The correlation between extent of resection and prognosis of a particular tumor type is of considerable importance. Historically, biopsy with irradiation was the standard of treatment due to patient morbidity and mortality with surgery. However, in low-grade thalamic tumors, radical resection can be curative. Consensus within the literature for low-grade astrocytomas includes stereotactic biopsy to verify histology, followed by surgery for curative intent. In a study examining the mortality rate of complete resection of benign thalamic astrocytomas in 26 children, the procedure was found to be associated with improved prognosis when postoperative MRI showed no residual tumor but was associated with a 7.7% mortality rate.37 Surgical resection of bilateral infiltrating astrocytomas of the thalamus is difficult to achieve, and biopsy for histologic diagnosis is preferred. These patients have a poor outcome despite adjuvant radiation and chemotherapy.
Anatomic considerations are important because the thalamus is adjacent to critical structures. The thalamus is bound anteriorly by the foramen of Monro and posteriorly by the posterior commissure. It also is bound superiorly by the stria medullaris and inferiorly by the hypothalamic sulcus. Finally, the thalamus is medially bound by the third ventricle and laterally bound by the internal capsule. The terminal vein lies in the groove between the thalamus and the caudate and serves as a landmark for the position of the internal capsule.34 Factors that must be considered by neurosurgeons include MRI appearance of the tumor, extent of tumor involvement, regions of enhancement, evidence of hydrocephalus, and relationship of the tumor to vital vascular structures.34 Surgical approaches that have been used with endoscopic or stereotactic guidance include transcallosal–interhemispheric, infratentorial–supracerebellar, and trans-sylvian transinsular approaches.34 Surgical resections can be performed while minimizing morbidity using concomitant neuronavigation and intraoperative neuromonitoring of the corticospinal tracts.
If the patient is in extremis because of elevated intracranial pressure secondary to hydrocephalus, the surgeon should perform emergent CSF drainage. The most rapid method is to place an extraventricular drain (EVD). If surgery for gross total resection is planned, the surgeon can attempt to wean the EVD postoperatively because the normal CSF flow is sometimes restored. If only biopsy is planned or a CSF blockade persists, the surgeon can perform an endoscopic third ventriculostomy or place a ventriculoperitoneal shunt.38
Postoperative imaging is obtained to evaluate the extent of resection and residual tumor. Adjuvant radiation and chemotherapy for high-grade gliomas are required. In a study of 57 patients, despite surgical resection and multimodal therapy, the median survival length remained 73 weeks.33 In contrast, children who have low-grade gliomas that have been resected in their entirety can be observed serially for tumor recurrence. Upward of 90% 10-year survival rates have been reported for low-grade thalamic gliomas. For partially resected or unresectable low-grade thalamic tumors, the timing of additional therapy is controversial. Most surgeons advocate a “wait and see” approach. Often adjuvant therapy can be delayed until signs of clinical worsening or radiographic growth. In one series looking at 128 children with subtotal resection of low-grade gliomas, 58% had no evidence of tumor progression 7 years after diagnosis despite undergoing no additional therapy. Furthermore, there was no difference in survival in those children that did receive immediate postoperative radiotherapy and those that not.39
Given the increasing data regarding the detrimental effects of radiation on children, many centers recommend chemotherapy as frontline adjuvant therapy in children with progressive low-grade gliomas. One combination that has shown promise is a combination of carboplatin and vincristine, which has been shown to generate a 68% 3-year progression-free survival rate.40 Unfortunately, 40% of children experience hypersensitivity reactions with carboplatin. These patients are often started on a regimen of 6-thioguanine, procarbazine, lomustine, dibromodulcitol, and vincristine, which has shown a 45% 3-year progression-free survival rate.40
Pediatric Glioblastoma
Glioblastoma multiforme (GBM) are WHO grade IV gliomas, which are rarely diagnosed in the pediatric population but cause significant morbidity and mortality. While GBMs are primarily an adult-age tumor, pediatric GBMs are also classified as high-grade gliomas, along with anaplastic astrocytomas and intrinsic pontine gliomas.41,42 Anatomically, the majority of these tumors are located supratentorially, but GBMs also occur in the brain stem.43 The current treatment recommendations include aggressive surgery, radiotherapy, and chemotherapy.
Epidemiology and Prognosis
Pediatric GBMs compose 3% of childhood primary brain tumors and are rarely diagnosed among children and adolescents. Overall, the prognosis of glioblastoma is better in children than in adults.44 The majority of patients experience recurrence after surgical resection within 2 years of diagnosis, with the overall survival rate at 67% 1 year, 52% 2 years, and 40% 5 years after diagnosis.44,45 The median overall survival length is 43 months, with a median progression-free survival length of 12 months.44 Survival is correlated with tumor location, and patients with superficially located GBM had a median survival length of 52 months, while those with deeply located tumors had a median survival length of 7 months.44 Furthermore, there is no significant relationship between patient age and survival.45 Histologic features, while prognostically significant in adults, have been found to have no association with survival in pediatric patients.45 Extent of resection was found to be a significant predictor of outcome. Radical tumor resection, defined as resection of greater than 90% of the tumor by postoperative imaging, was found to have a survival advantage when compared to partial resection in pediatric patients.42 Specifically, the median survival length for patients with completely resected tumors was 106 months, while those with incompletely resected tumors had a median survival length of 11 months.44
Pathology
GBMs are classified as WHO grade IV infiltrating astrocytomas. Pathologic diagnosis is based on histologic features that include pleomorphism, mitotic figures, endothelial proliferation, and tumor necrosis.46 Unlike adults, histologic grading has not been found to have a strong prognostic significance for children, as the difference in overall progression-free survival of pediatric glioblastoma and anaplastic astrocytoma is not statistically significant.47 Individual genetic alterations such as epithelial growth factor receptor (EGFR) amplification, p53 mutations, retinoblastoma loss, IDH1 mutations, and deletion of phosphatase and tensin homology (PTEN) are common in adults; however, such alterations are distinctly different in pediatric GBMs.46 In pediatric GBMs examined in the Children’s Cancer Group cohort of 945 patients, EGFR, PTEN, and IDH1 gene expression aberrance were rare in pediatrics, while p53 alterations were more frequent.47,48 In addition, overexpression of O-6-methylguanine-DNA methyltransferase, which is an important mechanism of resistance to temozolomide, is limited in pediatric glioblastomas and has driven investigation of the use of temozolomide in children.49 Pediatric GBMs are molecularly distinct from their adult counterparts and warrant therapies specifically directed toward these molecular targets.
Diagnosis
Presenting signs and symptoms vary by location and age. The history may include a short evolution of symptoms on the order of days and weeks to months.42 Cortical lesions may be manifested through seizures, hemiparesis, visual deficits, and headaches. Indications of obstructed CSF leading to hydrocephalus include morning headaches, vomiting, and papilledema.42 Infants commonly present with nonspecific symptoms, including irritability, lethargy, vomiting, failure to thrive, and macrocephaly. Nonspecific symptoms coupled with a neurologic deficit are particularly suspicious for a brain lesion in young children.
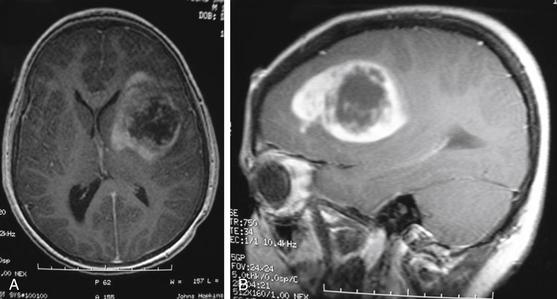
MRI is the preferred diagnostic imaging modality to determine whether symptoms are consistent with high-grade glioma. Imaging including axial, sagittal, and coronal planes pre- and postgadolinium should be obtained. T1 sequence characteristics include irregular margins, enhancement with contrast, invasion of surrounding brain parenchyma, heterogeneous enhancement patterns, and areas of cystic necrosis with a rim of enhancement (Fig. 56-3). Edema is commonly evidenced on FLAIR or T2. When imaging is consistent with high-grade glioma, biopsy—at a minimum—is indicated for tissue diagnosis. Stereotactic biopsy is appropriate for deep-seated tumors in functionally important areas.42 Further surgical management, including open resection, is dictated by tumor location.
< div class='tao-gold-member'>
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
Role of Gamma Knife Radiosurgery in the Management of Arteriovenous Malformations
 Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


