Chapter 133 Surgical Management of Severe Closed Head Injury in Adults
Additional text and figurees are available on Expert Consult
Traumatic brain injury (TBI) is defined as damage to the brain resulting from external mechanical forces. Head injury is the term used to define any alteration in mental or physical functioning related to a blow to the head. All TBIs are head injuries, but the latter is a broader term because it also refers to damage to structures other than the brain, such as the scalp and skull. However, the terms head injury and brain injury are often used interchangeably. The most widely used method for the assessment and stratification of TBI severity is the Glasgow coma scale (GCS), which categorizes patients according to the degree of impairment of consciousness, estimated from their verbal, motor, and eye-opening responses to external stimuli.1 Following this classification system, a severe head injury is defined by an initial postresuscitation GCS score between 3 and 8 points or by deterioration to a GCS score of 8 or fewer points after injury.2
Severe TBI represents a serious medical and scientific challenge. One of the central concepts that emerged from research is that all cerebral damage from TBI does not occur at the moment of the impact but rather evolves over the ensuing hours and days. Focused protocol-driven management, including rapid resuscitation and early intubation in the field, direct transport to a major trauma center, and improved critical care management in the hospital with intracranial pressure (ICP) monitoring and immediate evacuation of mass lesions, has cut mortality in severe TBI from as much as 50% in the 1970s to 15% to 25% in the 2000s. However, this tremendous advance seems to have plateaued in the last decade despite extensive experimental and clinical research about the cellular and molecular mechanisms involved in the progression of traumatic brain damage. Interventions using drugs to reduce the effects of mediators of injury have been encouraging in animal models, but clinical TBI trials have been ubiquitously unsuccessful so far. It is naïve to expect that severely head-injured patients, each showing a huge combination of different neuropathologic post-traumatic lesions at different stages of the disease, can all benefit from a single kind of treatment.3
TBI treatment is a team effort integrated by surgeons and nonsurgeons. The role played by neurosurgeons in the integral treatment of severely head-injured patients remains critical, especially regarding the appropriate therapeutic decisions to be made in advance to the progression of secondary brain damage. For a long time, the surgical management of TBI has been based on clinical anecdotal practice. The new paradigm of evidence-based medicine has broadened to the establishment of scientific surgical and medical guidelines, which should help neurosurgeons make the better decisions using a strong scientific background, in addition to their personal anecdotal experience.4,5
This chapter focuses on the physiopathology and surgical treatment of severe TBI. Besides providing the latest update on the evidence-based medical literature, a major objective of this chapter is to present a comprehensive review of the fundamental structural and functional properties of brain tissue, which is essential to understand the mechanisms underlying traumatic brain damage. The expanding advance of neurosciences in the last decades has produced outstanding insights in the intrinsic, dynamic mechanisms of cell damage set in motion with a head trauma. We firmly believe that succinct knowledge of these basic mechanisms can help neurosurgeons involved in the care of severely head-injured patients with better assessment of the complex clinical and diagnostic information obtained from each case. Furthermore, this knowledge represents the keystone for identifying potential future therapeutic targets for TBI. In the online version of this chapter, a section focusing on the neuroscience of TBI is provided.
Epidemiology
TBI represents a serious health problem and constitutes a major cause of death and severe disability in young and adult people in Western countries. TBI afflicts 1.4 million people yearly in the United States, resulting in 50,000 deaths and 235,000 hospitalizations.6 TBI is the number one cause of coma and the leading cause of brain damage in children and young adults. The incidence (number of new cases) of head injury is approximately 300 per 100,000 people per year (0.3% of the population), with a mortality of 25 per 100,000 people in the United States. Approximately 500,000 head injuries requiring admission to a hospital occur annually in the United States, and 50,000 people with these injuries die before reaching a medical facility. Of the 450,000 initial survivors, 80% sustain minor injuries (GCS score 13-15 points), 10% have moderate injuries (GCS score 9-12 points), and 10% have severe brain injuries (GCS score 3-8 points).6 Around 100,000 patients per year may require surgical management of a post-traumatic intracranial hematoma in the United States alone.4 Roughly 50,000 head-injured patients per year will have some form of permanent disability.7
Classification Schemes for TBI
The heterogeneity of TBI is considered one of the most significant barriers to finding effective therapeutic interventions. In addition, the lack of a valid classification system for TBI that is able to identify subgroups of patients who may survive with a reasonable neurologic outcome and thus may benefit from specific therapies contributes to this therapeutic failure. A great heterogeneity of pathoanatomic lesions can be observed in neuroradiologic studies of patients classified under the same “severe TBI” category; thus, it is difficult to conceive a general therapy able to target such variability of traumatic lesions.8 To review the most common types and patterns of injury associated with TBI and develop a classification scheme that would cluster TBI patients into groups based on their major types of injury, the National Institute of Neurological Disorders and Stroke sponsored a workshop on classification of TBI for targeted therapies in October 2007.2 On the premise that TBIs with similar pathoanatomic features are likely to share common physiopathologic mechanisms, a major conclusion from the workshop was that a new, multidimensional classification system for TBI should be developed and used in future TBI trials.
TBI Classification Systems for Targeted Therapies
Different classification systems for TBI have been used depending on the categories of the data chosen for the research. Head injuries have more often been distinguished using symptom classifications, which differentiate the patients according to the severity of their symptoms. However, pathoanatomic classifications could be more useful because they provide an accurate description of the lesions that should be treated. Finally, prognostic classifications describe the combination of factors associated with the patient’s outcome. Major disadvantages of classification systems are that the optimal time to classify patients is unknown and depends on the purpose of classification. Furthermore, personnel administering classification systems need to be trained to classify consistently and correctly. In general, the complexity of the physiologic derangement is the most important factor to take into account for triage, whereas the type of injury is of predominant importance when classifying patients regarding the selection of treatment. However, outcome-based systems of classification depend less on acute physiologic changes and more on functional capacity and permanent disability.2
Classification Schemes by Injury Severity
The most commonly used severity scale for TBI is the GCS, originally designed by Teasdale and Jennett to describe unconsciousness after brain injury (Table 133-1A).1 This scale includes assessment of eye opening, best verbal response, and best motor response. The total score, ranging from 3 to 15 points, should be ascertained after resuscitation, because the neurologic examination can be markedly altered by hypotension or hypoxia. The GCS is the most commonly used neurologic injury severity scale for adults because of its high interobserver reliability and generally good capability of predicting the outcome. However, the main disadvantages of the GCS system are that summation of assessment scores results in loss of information, because multiple combinations of eye, motor, and verbal scores can equal the same total score, and that pupillary reaction and lateralization of the exam are not included. Furthermore, the reliability of this score is questionable in the setting of sedative and paralytics use and the optimal timing of measurement is unclear, with a change in GCS score being more relevant than a single measure. TBI patients are typically divided into three subgroups based on the total score: severe TBI (GCS score 3-8 points), moderate TBI (GCS score 9-12 points), and mild TBI (GCS score 13-15 points). Most severely head-injured patients are in a coma, which is defined by the International Coma Data Bank as the inability to obey commands, utter words, or open eyes.9

Classification Schemes by Pathoanatomic Findings
Schemes of classification by pathoanatomic findings describe the location and type of abnormalities to be targeted by treatment. The majority of severely head-injured patients have more than one type of injury when classified in this way, such as scalp lacerations, skull fractures, intracranial hematomas, traumatic subarachnoid hemorrhage (SAH), intraventricular hemorrhage, cerebral contusions, and diffuse patterns of injury such as diffuse axonal injury (DAI), diffuse brain swelling, or brain ischemia. Each of these entities can be further described by their location, extent, multiplicity, and so on. Gennarelli and Thibault were among the first authors to incorporate both GCS and computed tomography (CT) findings to the evaluation of the outcome after TBI, differentiating patients on the basis of the type of lesion showed by CT (focal or diffuse), their GCS score, and the length of their coma.10 They found that subdural hematomas (SDHs) and diffuse injuries associated with a coma for more than 24 hours had the highest mortality. Lobato et al., after a thorough evaluation of CT lesions displayed by patients with severe TBI, also concluded that SDHs, in addition to multiple brain contusions, were associated with the highest mortality.11
In 1991, Marshall et al. described a CT classification system based on information gathered from severe TBI patients in the Traumatic Coma Data Bank (TCDB).7 This scheme is based on the appearance of perimesencephalic cisterns, the presence of midline shift, and the presence of focal masses, and it classifies CT findings into six groups (Table 133-1B). Among the different pathoanatomic classification schemes proposed, this has been shown to be the most successful in predicting both risk of increased ICP and outcome in adults. Diffuse injury type IV was associated with the highest mortality. Maas et al. evaluated Marshall et al.’s classification system in comparison with the use of individual CT findings for prognosis after TBI and observed some discrepancies in classification, with up to 30% of patients classified as having diffuse injury type IV being reported without a midline shift greater than 5 mm (the basic criterion of diffuse injury type IV).12
Table 133-1B Classification by Pathoanatomic Findings (Marshall’s Computed Tomography Classification7)
| Type | Radiologic Findings on CT |
|---|---|
| Diffuse type I (no visible pathology) | No visible intracranial pathology seen on CT scan. |
| Diffuse type II (no swelling and no shift) | Cisterns present with midline shift < 5 mm and/or lesion densities present. No high- or mixed-density lesion > 25 ml. May include bone fragments and foreign bodies. |
| Diffuse type III (swelling) | Cisterns compressed or absent with midline shift = 0-5 mm. No high- or mixed-density lesion > 25 ml. |
| Diffuse type IV (shift) | Midline shift > 5 mm. No high- or mixed-density lesion > 25 ml. |
| Type V (evacuated) | Any lesion surgically evacuated. |
| Type VI (nonevacuated) | High- or mixed-density lesion > 25 ml not surgically evacuated. |
Classification Schemes by Biomechanical Mechanism
Injuries can be classified according to whether the head is struck or strikes an object (contact or impact loading) and/or the brain moves within the skull (noncontact or “inertial” loading). The magnitude and direction of the combined loading forces may predict the type and severity of injury.10 There is considerable but not perfect correlation between physical mechanism of injury and pathoanatomic injury type. For instance, most focal injuries, such as skull fractures, brain contusions, and epidural hematomas (EDHs), result from impact loading, whereas inertial loading generally causes more diffuse injuries, such as concussion, SDHs, and DAI. However, in clinical practice, mechanistic classification is difficult to apply because of the uncertainty about the loading conditions involved in the TBI.
Classification Schemes by Pathophysiology
Pathophysiology classification schemes include biochemical, metabolic, neurophysiologic, and/or genetic dynamic processes set in motion by the injury. One widely used physiopathologic scheme in TBI was introduced by Adams et al., who differentiated between primary injury, or unavoidable, immediate-impact parenchymal damage, and secondary injury, or potentially avoidable damage.13 However, the importance given to secondary insults has gained recognition, and since the 1990s, understanding and prevention of delayed damage have received priority. Secondary lesions are defined as those set in motion by the impact but not pathologically or clinically manifested until minutes, hours, or days after the injury. In addition to secondary ischemic injury, probably the most relevant factor influencing a worse outcome, secondary damage can be caused by neurotransmitter dysfunction, cellular and extracellular edema, blood–brain barrier (BBB) disruption, mitochondrial damage, genetic alterations such as apoptotic induction, and so on.14
Classification Schemes by Prognostic Modeling
Outcome after TBI is multifactorial and best assessed with a multidisciplinary approach. Outcome mainly depends on severity of injury, comorbid conditions, preinjury status, social factors, and patient age. The ideal TBI classification system should be able to select patients who potentially could benefit from a considered therapy from a clinical, pathoanatomic, physiopathologic, or prognostic perspective. Traditionally, the Glasgow outcome scale (GOS) has been used to categorize the long-term prognosis of patients after TBI, and the GOS score has been dichotomized into 3 to 5 points versus 1 to 2 points (Table 133-1C). Although the 5-point GOS score has been criticized for lacking sensitivity and for having a ceiling effect, this is still the most widely used scale for classifying outcome after TBI. Most TBI patients fall into the extremes of the scale, good recovery or death, and the scheme has undergone several revisions and was extended to an 8-point scale to address some of these limitations. Both the GOS and the extended GOS have been extensively used and have been shown to have validity and reliability in measurement of outcome by trained staff. Both scales are also correlated with other measurements of disability, mental status, and neurobehavioral functioning. Recent work from the International Mission for Prognosis and Clinical Trial has resulted in the development of three valid prognostic models of increasing complexity, a task particularly relevant to identifying subgroups of patients likely to benefit from a specific therapy in phase III trials.8
Table 133-1C Classification by Prognostic Modeling (Glasgow Outcome Scale)
| Score | Features |
|---|---|
| 1 | Dead |
| 2 | Vegetative state (unable to interact with environment, unresponsive) |
| 3 | Severely disabled (able to follow commands, unable to live independently) |
| 4 | Moderately disabled (able to live independently, unable to return to work/social activities) |
| 5 | Good recovery (patient has resumed most normal activities) |
Biomechanics of TBI
TBI is generally the result not of a direct impact to the skull but of inertial linear and rotational acceleration/deceleration (A/D) forces acting on brain tissue and causing structural damage and functional disturbances on the different types of brain cells. This fact was discovered early in the history of TBI research by Denny-Brown and Russell, who observed that the loss of consciousness (LOC) associated with concussion was difficult to inflict when the movement of head during the application of the impact was restricted.15 Nervous tissue strains induced by both linear and rotational forces create spatiotemporal deformation gradients (Fig. 133-1). Because the brain is a viscoelastic organ with little internal structural support, it poorly tolerates such forces. Gray matter closest to the surface of the brain is most susceptible to linear A/D forces. In contrast, the deeper cerebral white matter axons and gray matter of deep nuclei, in addition to the axonal tracts within the brain stem, are more vulnerable to rotational A/D forces. In the early 1940s, the effects of A/D forces on the brain were assessed on a physical gelatin model resembling gross brain’s physical properties, such as its volume, mass, and deformability.16 These studies were the first to describe how shear strains were caused by rotational A/D forces (also known as impulsive loadings) (Fig. 133-2). One decade later, the effects of A/D forces on brain tissue were analyzed by exhaustive postmortem neuropathologic studies performed on the brains of patients who had died from lethal road accidents.17 These studies showed that the predominant microscopic finding was diffuse degeneration of white matter characterized by the development of axoplasmic “retraction balls” at the sites of axonal discontinuity, without obvious damage to the cortex. The term that Strich used to define this damage was “shearing injury,” because he thought that traumatic damage to white matter was caused by the shearing of nerve fibers at the moment of injury.17 Clinicopathological studies undertaken by Adams et al.13 endorsed Strich’s views, and Jennet and Plum considered this to be the most common cause of the vegetative state (VS) observed in many severely head-injured patients.9 In patients with shorter survival times (less than 6 weeks following the injury), the specific microscopic pattern of axonal degeneration consisted of the presence of retraction balls, interpreted as end swellings of proximal and distal segments of severed axons, which axoplasm had extruded.18,19
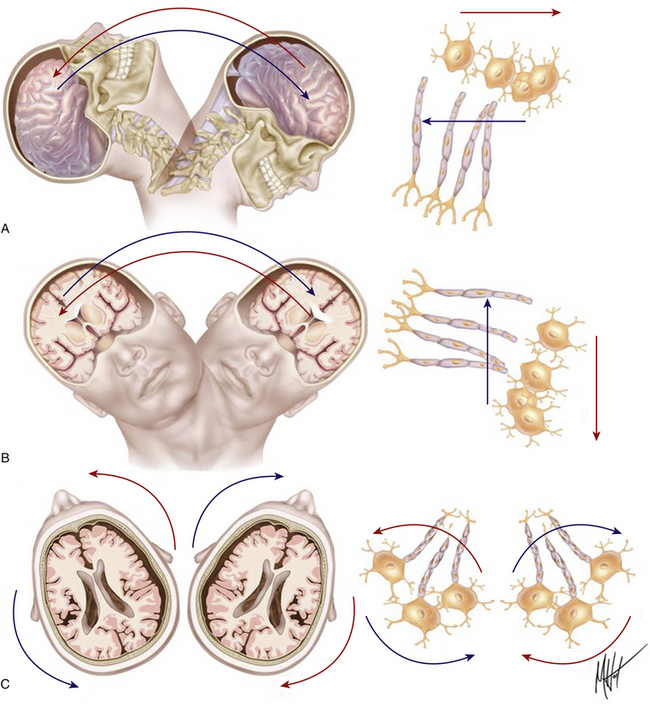
FIGURE 133-1 Biomechanics of traumatic head injury I. Classic model of axonal shear injury caused by rotational A/D forces. Impulsive inertial loadings cause damage to axons grouped in bundles within white matter without direct impact to the skull. The severity of axonal injury depends on the course followed by the fibers and the predominant spatial plane of action of the rotational A/D force: sagittal (A), lateral/coronal (B), or axial (C). Different tissue densities of gray and white matter determine differences in the inertial displacement experienced by both tissues, causing maximal stress on a definite segment of the axons. Rotational A/D forces in the lateral plane cause the severest damage to the long axonal tracts coursing along the brain stem.18,20
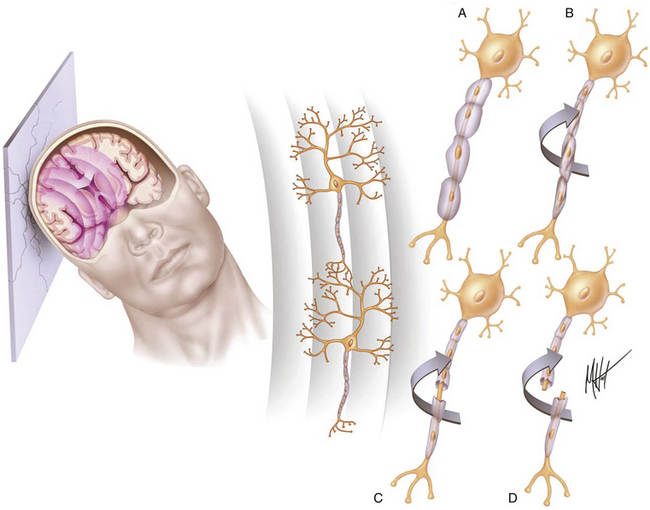
FIGURE 133-2 Biomechanics of traumatic head injury II. Classic model of spatiotemporal deformation gradient caused by A/D forces. Impact loadings inflicted on the freely moving head initiate a deformation wave that travels in the viscoelastic brain tissue, creating a spatiotemporal gradient.15,16 The major structural lesion is characterized by a stretching injury of the axolemma of scattered neurons, which cause development of multifocal swellings (A) and blocking of normal axonal transport (B), but the maximal degree of deformation may cause direct tearing (C) or breaking (D) of axons.35
Diffuse brain injury usually depends on inertial loadings as a result of rapid head rotational motions of the brain, resulting from unrestricted head movement in the instant of injury.20 Rapid A/D head movements deform white matter, inducing dynamic shear and tensile and compressive strains within the tissue (Figs. 133-1 and 133-2). A significant theoretical insight regarding the biomechanical effects of A/D forces on brain structure was made by Ommaya and Gennarelli in 1974.21 These authors proposed a “centripetal model” of severity of structural damage known as the Ommaya-Gennarelli model of TBI, which was based on experimental studies performed in subhuman primates.20,21 This probably represents the most influential biomechanical model in the field of head injury, because it explains the damage caused by A/D forces, which are most common in traffic accidents. According to this model, the mechanical strain injury operates in the brain tissue in a “centripetal sequence,” in which the higher the magnitude of the injuring forces, the deeper the brain functional and structural damage observed. The sequence of damage begins at the surface of the brain and progressively affects deeper structures as the A/D forces increase in intensity. This model uses the principle that the severity of injury is mainly determined by the direction of force, with rotational forces causing the severest injuries. On the basis of the centripetal progression of injuries, the model introduced three critical predictions: (1) when the degree of trauma is sufficient to cause LOC, cortical and subcortical structures are primarily affected and show more severe damage than that found in the rostral brain stem; (2) damage to the rostral brain stem does not occur without severer damage of cortical and subcortical structures, since the mesencephalon is the deepest structure and the last one to suffer the effects of trauma; and (3) cognitive symptoms such as confusion and disturbance of memory can occur after TBI without LOC; however, the reverse cannot occur.
The Ommaya-Gennarelli centripetal model challenged the sustained view that the main mechanism producing traumatic unconsciousness after TBI was an isolated brain stem injury. The primary brain stem damage theory was supported in the 1950s and 1960s, when the ascending arousal system (AAS) was described by Moruzzi and Magoun.22 Experimental models of TBI on subhuman primates in which the head of an animal was exposed to A/D forces using accelerator devices showed that nonimpact A/D caused concussion and axonal injury only when the moving head was allowed to rotate, while linear accelerations induced gross structural damage but not DAI.20,21 These findings were later confirmed in clinical studies, in which A/D loadings to the head in the coronal plane were observed to cause the severest injuries in humans subjected to motor vehicle accidents. Although early studies suggested that the primary site of brain injury in patients presenting with transient LOC and post-traumatic cognitive deficits was the brain stem, overwhelming neuropathologic evidence showed that most mild and moderate TBI caused damage to widespread areas of the cortex and the subcortical white matter.13,23 Recent magnetic resonance imaging (MRI) studies, in which the assessment of DAI was performed during the first month after injury, confirmed the Ommaya-Gennarelli model.24
Biomechanical theories involving linear and rotational A/D loadings are considered by many authors to be incomplete and may not adequately explain injury to deeper cerebral structures in the absence of injury to more superficial cerebral structures. Early biomechanical work suggested that brain injuries occurred principally from pressure gradients developed within the brain at the time of the impact (Fig. 133-2), but more recent research has shown that head motions occurring at the moment of injury result not only in pressure waves but also in intracranial brain deformations, which are thought to be a primary cause of the post-traumatic brain lesions. The stereotactic theory considers brain tissue to be a spherical, deformable structure composed of concentric planes with the same density in which skull vibrations generate secondary pressure waves that propagate as a spherical wave front. The spherical nature of this wave front is able to focus its energy on deeper cerebral structures without an associated injury at the brain surface.
Historical Studies on TBI: Pioneering Research By Ramón y Cajal
The Nobel Prize winner Ramón y Cajal (1852-1934) was a distinguished pioneer in the scientific study of the histologic consequences of traumatic injuries on brain tissue (Fig. 133-3A).25 He performed experimental studies on the brains of dogs and cats after severing the brain tissue with a scalpel and then studied the effect of the injury at different times up to 1 month following the injury. Ramón y Cajal described for first time the phenomenon of preservation after traumatic injury, which consists of the necrosis of cell elements distal to the lesion while these elements display perfect morphologic integrity. He also described for the first time the terminal clubs and hypertrophic stumps developing at the proximal endings of the surviving axons (retraction balls) and at the distal stumps of the preserved detached axonal segments (Fig. 133-3C).26 Retraction balls were evident in the damaged tissue 24 hours after injury, and some thin axonal appendices were seen expanding out from each ball in the direction previously followed by the axon. In addition, 6 to 8 hours after injury, he observed varicosities along the fibers proximally to the retraction balls. Retraction balls and varicosities were observable during approximately the first week after the injury and then underwent a process of degeneration. Later, and nearer the cell bodies, new buds formed (late retraction balls). These persisted for 2 to 3 weeks and then disappeared, with some perhaps entering into the formation of the last living axon collateral, which became markedly hypertrophied.25
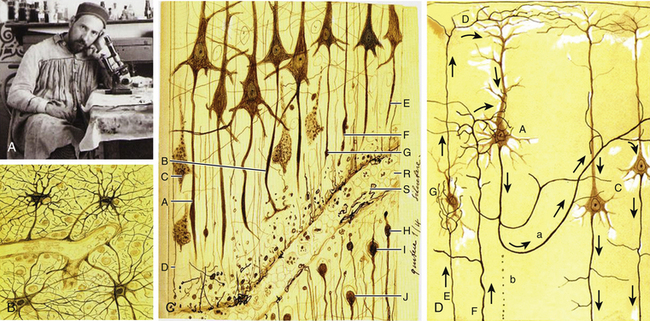
Therefore, the cortical pyramidal cells, which are those emitting long axons, were transformed after the traumatic injury into cells with short axons and hypertrophied collaterals (Fig. 133-3D). Ramón y Cajal proposed that the collateral hypertrophy is a compensatory phenomenon similar to the effects of an arterial ligature. Nevertheless, he did not know the fate reserved for the pyramidal cells with short axons. Months after the injury, he could not observe retraction balls or varicose fibers near the lesion; rather, he saw only smooth well-stained axons presenting all the signs of being normal conductors. He also observed that central tracts were incapable of regeneration and that although they had the property of producing new fibers, once the reconstructive movement is initiated, for unknown reasons the nerve sprouts lose their energy and suspend their growth. Nevertheless, he described neoformations (sprouting) in the pyramidal axons that were maximal in the second or third day after injury. However, in a few days these neoformative axon structures disappeared, and after 15 days, they were absent. In addition, he described the morphologic reactive changes in glial cells after the injury, consisting of the loss of capillary–neuroglial relationships (dislocation of perivascular astrocytes) and free migration of these cells to distant regions.25
Neuropathology of TBI
In 1990, Adams summarized the neuropathologic postmortem findings obtained from one of the largest series of TBI patients providing a full postmortem examination. These series were part of the Glasgow database, which consisted of 635 fatal closed head injuries gathered between 1968 and 1982 in Glasgow, including 434 patients with a detailed neuropathologic study.18 One third of these patients had “talked” at some time after the injury.27 The first major conclusion from this work was that severe brain damage can be caused without anything striking the head or the head striking anything. The second conclusion was that DAI represents the most important factor influencing the outcome in anyone who sustains a nonmissile head injury followed by secondary brain ischemia, which represented the second prognostic factor in importance after TBI.
According to the pathologic classification introduced by Gennarelli in 1987,28 two major types of post-traumatic lesions could be differentiated in Adam’s series18: focal lesions, which are large enough to be identified by the naked eye, including cerebral contusions and intracranial hematomas, and diffuse injuries, which are all that cannot be macroscopically identified, including DAI, diffuse brain swelling, secondary hypoxic/ischemic brain damage, and diffuse vascular injury (Figs. 133-4 and 133-5). Overall, cerebral lesions caused by TBI can be viewed as a spectrum of injuries ranging from purely focal to purely diffuse, all brought about by A/D forces causing mechanical distortion to the head and nervous tissue. Focal brain injuries are usually produced when the stationary skull is struck by a moving object with relatively small mass, such as a stick or baseball bat. Focal brain injuries are characterized by a central area of structural disruption, surrounded by an area of primary traumatic damage without destruction of brain tissue, and finally enclosed by a peripheral tertiary zone of potential delayed insult associated with ischemia and edema.
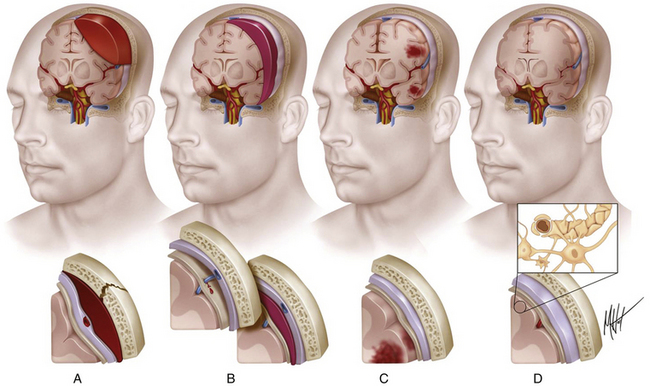
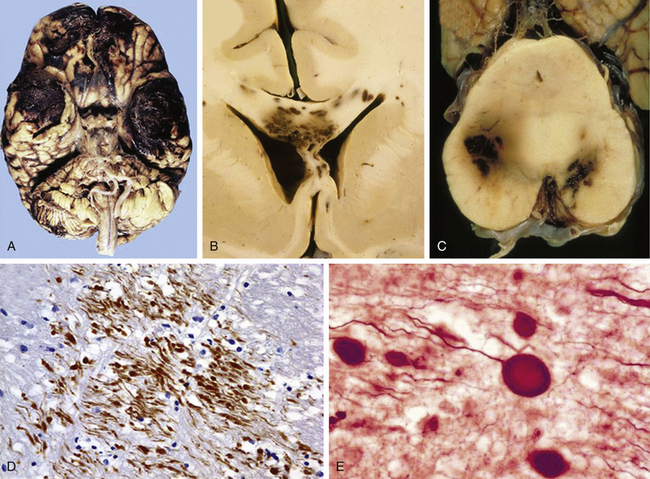
The importance of diffuse brain lesions caused by TBI was first emphasized by Symmonds as the cause of initial LOC and the frequent, long, continued disturbances of consciousness, often followed by residual symptoms in patients who were rendered unconscious at the moment of injury.14a Later experimental work by Denny-Brown and Russell15 and more recent work by Gennarelli et al.20 linked this type of damage to the prolonged unconsciousness occurring in almost half of severely head-injured patients in the absence of intracranial mass lesion. Diffuse injury is difficult to define during life, because much of it can only be recognized microscopically. Even in postmortem studies, it can hardly be recognized unless the brain is properly fixed prior to dissection. Nevertheless, this type of injury should be suspected in patients in a coma who do not present focal lesions.
Intracranial Hemorrhages
Intracranial hemorrhagic lesions represent the most common cause of clinical deterioration and death in patients who experience a lucid interval and usually develop in patients sustaining a skull fracture.27 If the lucid interval is longer than 24 hours, the hematoma is said to be “delayed.” This occurs in 30% of head-injured patients with intracranial bleeding due to the development of a new hematoma after an initial normal CT scan. The three most important intracranial hemorrhagic lesions caused by TBI are EDHs, acute SDHs, and ICHs (Fig. 133-4A-C). EDHs corresponded to the 8% of lesions observed in the Glasgow database, whereas “pure” SDHs were diagnosed in 13% and ICHs in 15% of lesions. Nevertheless, a higher rate of 26% of patients presenting with SDHs associated with ICHs was observed in the context of a “burst lobe.” This term describes a massive lobar lesion showing the coexistence of cerebral contusions, blood in the subdural space, and a hematoma in white matter deep to contusions (Fig. 133-5A). In a study performed in 1987 comparing 100 fatal head injuries with cerebral hematoma and 100 without cerebral hematoma, the cases with hematoma were observed to affect predominantly older patients.29 Structural factors such as parenchymal atrophy and weaker holding of intracerebral small-caliber vessels may probably account for this observation. Other less frequent types of focal brain injuries are traumatic lesions of the corpus callosum, usually associated with DAI; pontomedullary rents or tears in the brain stem at the junction between the pons and the medulla, often associated with a ring fracture of the base of the skull; lesions of the hypothalamus and the pituitary stalk; and lesions of the cranial nerves, with loss of sense of smell sometimes occurring after head injury because of avulsion of the olfactory nerves. For a detailed description of intracranial hematomas, refer to the section on surgical treatment of TBI.
Cerebral Contusions: Major Gray Matter Focal Lesions After TBI
Brain contusions represent frank disruptions of brain tissue, which are typically observed at the apex of cerebral gyri, appearing as coalescent petechial hemorrhages or streaks of hemorrhage with an eventual progression of bleeding into adjacent white matter (Figs. 133-4C and 133-5A).30 Brain contusions and lacerations are considered primary TBI lesions that are observed at the poles and orbital surface of the frontal lobes in up to 80% of autopsy studies performed on patients dying after the trauma (Fig. 133-5A). These are the brain areas undergoing the strongest inertial impact against the rough and irregular prominences of the inner surface of the skull following abrupt A/D forces. Contusions are basically impact-related lesions occurring with short-duration and high-acceleration impacts.20 Both brain contusions and lacerations are always accompanied by a certain degree of edema and by SAH, while intraventricular hemorrhage accompanies DAI in most cases. Cerebral lacerations represent destructive post-traumatic lesions in which disruption of cerebral tissue necessarily occurs, usually involving both the cortex and the subcortical area. Direct lacerations are caused by penetrating craniocerebral wounds, but indirect lacerations resulting from severe A/D shearing forces can be also observed.
Comprehensive studies performed by Lindenberg and Freytag in the 1950s34 and by Oprescu in the 1960s30 identified some specific types of contusions in the brains of head-injured patients dying a short time after the injury, such as contusions beneath skull fractures, coup contusions at the site of impacts (Fig. 133-4C), contrecoup contusions in regions distant to but not always opposite the impact site31 (Fig. 133-5A), herniation contusions, and finally, gliding contusions,32 which correspond to bilateral, focal, post-traumatic hemorrhages in the cortex and subcortical white matter of the superomedial parasagittal margins of cerebral hemispheres.30 Gliding contusions are thought to be caused by rotational A/D forces and are usually associated with DAI and small post-traumatic hematomas in the basal ganglia.
The complex dynamic sequence of the damage in cerebral contusions is not well understood. Early studies by Scheinker suggested that the cerebral vessels are primarily injured by the TBI and the parenchymatous tissue develop a progressive secondary damage.33 An initial vasodilatation followed by precapillary paralysis could be the triggering disturbance, causing endothelial leaking with blood extravasation that may lead to the development of true ICHs. In addition, the development of vascular thrombosis produces areas of necrosis that may become hemorrhagic, with a combination of necrotic and hemorrhagic brain tissue.30 The classic study of Lindenberg and Freytag identified the presence of two components in cerebral contusions: (1) the central area (core), in which cells undergo necrosis as the primary consequence of mechanical vascular and parenchymal injury, and (2) the peripheral area (rim), in which cellular swelling is observed.34 In contusive areas, neuronal bodies show vacuolar degeneration with chromatolysis and their axons show a varicose degeneration, a type of lesion originally described by a Ramón y Cajal in 1928 and later reported by Strich.13,17,25 Glial alterations are constantly seen with hypertrophy, swelling of astrocytes, and the development of reactive hypertrophic microgliosis or “microglial stars.” Up to 14 days after TBI, axonal retraction balls generally develop, rapidly followed by microglial and astrocytic reaction. From then on, a progressive wallerian degeneration of axons is clearly visualized.35
Physiopathology of Brain Contusions: The Osmotic Model of Progression
A rapid, massive type of cerebral edema associated with contusions may develop progressively within 12 to 72 hours after trauma, determining a marked high ICP and the shift of brain tissue, which may result in a delayed neurologic deterioration known as the “talk and deteriorate” syndrome.36,37 In addition, a “benign” slow form of delayed pericontusional edema, which rarely causes ICP elevation, is typically seen on T2-weighted MRI in white matter several days after injury.38 Contrary to the widely sustained view that increased cerebrovascular permeability is responsible for the development of contusion edema, some immunohistochemical and MRI studies have observed that vasogenic edema does not develop in contusions until 48 hours after injury, predominantly in white matter surrounding the lesion.39 Therefore, another mechanism should account for the cases in which rapid and massive contusion-associated cerebral edema develops.
In the central area of contusion, the cellular elements uniformly undergo mechanical disintegration and homogenization. This process can create a rapid increase in osmolality in the core of the contusion that attracts a large amount of water. Recent MRI–diffusion-weighted imaging (DWI) studies have observed that a large amount of edema fluid accumulates in the necrotic core of brain contusions up to 72 hours after trauma, resulting in rapid expansion of the extracellular space detected by an increased value of the apparent diffusion coefficient.38 Samples of necrotic brain tissue during surgical evacuation of cerebral contusions have revealed very high osmolality within the necrotic core of cerebral contusions when compared with the healthy brain tissue.40 Similar findings were observed in brain contusions induced in experimental TBI models. The total concentrations of Na+, K+, and Cl− are not significantly altered within the tissue, and the accumulation of metabolic intermediate osmoles (idiogenic osmoles)—which are produced in pathologic enzymatic activation, such as catabolism of proteins and lipids and deoxyribonucleic acid (DNA) fragmentation—might account for the hyperosmolar gradient created within contusions. An elevation in the brain tissue content of idiogenic osmoles associated with the formation of edema has been demonstrated in cerebral ischemia and in the early development of post-traumatic diffuse brain swelling in experimental TBI models.41
Natural History and Surgical Treatment of Cerebral Contusions
Despite intensive medical therapy, the elevated ICP caused by early massive edema associated with post-traumatic cerebral contusions is often uncontrollable and fatal. An explanation to this therapeutic failure would be that extremely high osmolality values (greater than 380 mOsm) reached within brain contusions make these lesions resistant to hyperosmotic diuretic therapy. Consequently, surgical treatment may be the best option for the efficient control of ICP. Nevertheless, indications for surgical intervention for severe cerebral contusions remain controversial.4 A recently published retrospective review of 98 patients harboring cerebral contusions who initially received conservative treatment showed that 45% of brain contusions displayed a significant progression on follow-up CT and 20% required surgical intervention.42 Contusion progression most likely happened in the first 2 days after injury, and no patient with an initial GCS score of 15 points or an initial contusion size of less than 14 ml required delayed evacuation.
The effects of surgical excision of the necrotic brain tissue in patients with severe cerebral contusions has been investigated in the Japan Neurotrauma Data Bank, in which a total of 1002 severe TBI patients were included.43 From 182 patients presenting with severe cerebral contusions, 66% were treated conservatively, whereas 34% underwent surgery. In most surgical cases, excision of necrotic tissue and clots, in addition to decompressive craniectomy, was performed. A poorer outcome was observed in the group treated conservatively 6 months after injury, which showed a double mortality rate (48% vs. 23%). These data support the usefulness of surgical treatment for cerebral contusions to prevent clinical deterioration and death, especially in patients with GCS scores of 9 points or better at the time of admission (patients who talk and deteriorate).
Basic Concepts of DAI
The term diffuse axonal injury, also known as diffuse white matter shearing injury, was coined by Gennarelli et al. to describe the scattered injury of white matter fibers observed throughout the brains of animals and humans who suffered TBI.20 DAI was observed in 30% of the cases included in the Glasgow database, typically in cases of head injury involving a pure inertial A/D noncontact loading of the head.18,44,45 Patients who sustain severe DAI are unconscious from the moment of the impact and do not experience a lucid interval, remaining unconscious, vegetative, or at least severely disabled until death. DAI has been observed in 80% of patients suffering from a VS. In the past, these severe alterations of consciousness after TBI were considered the exclusive effect of brain stem injury due to dysfunction of the fibers of the AAS, but although brain stem damage occurs in many cases, it is always accompanied by evidence of DAI. In patients recovering from a coma, DAI contributes to the functional neurologic disturbances usually present, such as spasticity, intellectual decline, and unmodulated behavior patterns.
Axonal damage is usually caused by forces occurring in less than 50 msec, as seen in automobile crashes, in which the brain tissue behaves as a stiff mass transmitting tensile elongation to axons that cause damage to their cytoskeleton. The first description of specific diffuse brain lesions in patients in a permanent VS after TBI until death was provided in 1977 by Adams et al., who identified a classic clinicopathologic pattern consisting of a triad of hemorrhagic lesions in white matter, corpus callosum, and rostral brain stem typically observed in the severest form of DAI.13 According to the severity of the axonal damage, Adams et al. proposed a classification of DAI in three categories of severity (Fig. 133-5B and C and Table 133-2).46 Nevertheless, some degree of diffuse axonal damage is likely to be present in all patients with severe head injuries and is almost universally present in fatal TBI.24

The fundamental microscopic, neuropathologic finding of DAI is the development of globular expansions developed at the proximal and distal stumps of severed axons known as axonal retraction balls, which were first described by Ramón y Cajal in 1911 in models of traumatic injury after causing injury to the cerebral cortex or spinal with a surgical knife (Fig. 133-3C and D).26 Nearly half a century later, Strich described the same neuropathologic finding in the brains of patients in a permanent VS after TBI and stressed that the disruption of white matter fibers was caused by shearing forces.17 For a long time, it was believed that tensile forces on axons caused a physical disruption of these structures, splitting them into proximal and distal segments, and that this disruption produced discharging of the axoplasm content to form a swollen proximal end known as a retraction ball or end bulb (Figs. 133-1, 133-2, and 133-5D and E).25 However, traumatic primary axotomy only occurs with the strongest shearing forces, and most axons exposed to A/D forces do not tear or break at the moment of the injury; rather, they undergo dynamic, progressive, structural and functional alterations that potentially may lead to a secondary axotomy without necessarily associating it to physical disruption of the axolemma.23 Recent evidence suggests that axons are far more resistant to stretching than previous studies indicated and human neurons in culture are able to tolerate tensile strains up to 65% of their original length without undergoing axotomy.
Diagnosis of DAI
The diagnosis of DAI can only be confirmed on postmortem studies using immunohistochemical methods such as antibodies against the beta-amyloid precursor protein (β-APP) or against the 68 KD neurofilament subunit. The β-APP is a protein normally transported along the axon that accumulates at the axonal beads after DAI and can be immunohistochemically detected, serving as an accurate marker for impaired axonal transport.23,47 In TBI, β-APP accumulates before disruption of the axolemma and apparition of axonal retraction balls occur, suggesting that axonal transport failure is an early event (eFig 133-16 A). Using these immunohistochemical methods, DAI is detected in nearly 100% of moderately and severely head-injured patients.
It is accepted today that patients in whom the CT scan discloses focal petechial hemorrhages represent only the severest forms of DAI and that MRI can show may more lesions than CT in a large number of these patients. Shearing injuries are displayed on MRI studies as multiple, small, ovoid lesions of 5 to 15 mm with their long axis paralleling the direction of the affected axons, most frequently found in the corpus callosum and in parasagittal regions of the brain.24 Intracranial hematomas can be observed associated with DAI, as well as brain swelling, in 75% of cases. The most recent MRI studies confirmed the three-stage classification scheme of DAI severity proposed by Adams et al., with only the severest stage III associated with a poor outcome (Fig. 133-5B and C and Table 133-2).24,48
Neuroradiologic Diagnostic Methods in TBI
CT Scanning
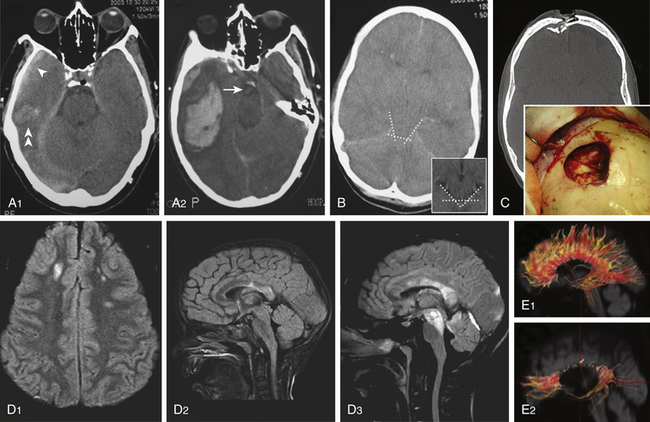
Cranial CT scanning is the imaging modality of choice for evaluation of a head-injured patient. It allows rapid assessment of the location, extent, and type of intracranial lesions caused by the head injury. With the lateral scout view and CT bone windows, skull fractures usually are well visualized, thus obviating the need for skull radiographic studies. Three major findings should be evaluated on a head-injured patient’s CT: (1) presence of intracranial hematomas, (2) appearance of basal cisterns at the midbrain level, and (3) presence of traumatic SAH. The status of the basal cisterns is related to the outcome. Basal cisterns can be open (all limbs are open), partially closed (one or two limbs are obliterated), or completely closed (all limbs are obliterated) (Fig. 133-6B). Compressed or absent basal cisterns indicate a threefold risk of raised ICP. Mortality is increased twofold in the presence of traumatic SAH, a sign that, when observed at the basal cisterns, carries a positive predictive value of unfavorable outcome of approximately 70%.4
Approximately 10% of initial head CT scans in patients with severe TBI do not show abnormalities, and significant new lesions associated with increased ICP may develop in 40% of patients with an initially normal head CT scan. A CT scan should be repeated, especially if the first scan was obtained within a few hours of injury or if the patient has the potential of developing surgical lesions such as cerebral contusions, before clinical signs of neurologic deterioration are apparent (Fig. 133-6 A1 and A2). A delayed or progressive brain injury, defined as new or progressive lesions seen on a follow-up CT scan, is observed in 45% of a large series of patients with closed head injury. McBride et al., based on a review of 154 consecutive patients with closed head injury requiring surgical intervention, recommended a repeat CT scan within 4 to 8 hours of the initial scan in all patients with an abnormal initial scan, given that nearly 50% of patients received surgical intervention based on the findings of the follow-up CT scan.49 Risk factors of developing delayed cerebral insults are the clinical severity of the head injury as defined by the initial GCS score, the need for cardiopulmonary resuscitation at the accident site, the presence of an acute SDH on the first CT scan, and the presence of coagulopathy on admission. Of those patients with delayed brain injury, 55% had abnormal clotting studies, whereas only 9% of those patients whose follow-up CT scans did not worsen had abnormal coagulation studies.
Magnetic Resonance Imaging
MRI is recommended in post-traumatic coma unexplained by CT scan, both for diagnosis and for assessing prognosis. MRI is a better prognostic tool for TBI than CT scanning because it can detect DAI and brain stem lesions. Use of MRI in the acute phase was initially restricted by practical issues, such as availability, compatibility with life-support equipment, and resource capacity. These issues have been addressed; consequently, an increasing number of studies have reported on the use of MRI in severe TBI. It has been observed that 85% of severely head-injured patients showing brain stem injury on MRI have an unfavorable outcome (death, VS, or severe disability) compared to only 55% of patients without brain stem lesions.48
Table 133-3 summarizes the most important characteristics and findings obtained with MRI sequences after TBI. A conventional T2-weighted MRI sequence is useful but limited in detecting lesions within the periventricular area or the cerebral cortex due to the presence of cerebrospinal fluid (CSF) nearby. The MRI–fluid-attenuated inversion recovery (FLAIR) sequence is sensitive in evaluating nonhemorrhagic lesions on white matter because it reduces or suppresses the signal produced by CSF, thereby detecting a larger number of lesions (Fig 133-6 D). Injured white matter volume within 2 weeks of injury (total DAI volume) has been observed to be correlated with patient outcome. MRI–DWI is more sensitive to DAI and can see alterations 3 hours after injury because it evaluates the altered local water diffusion. It distinguishes cytotoxic from vasogenic edema. Timing of MRI is important, because in DAI regions there is an evolution from restricted diffusion (cytotoxic edema) to unrestricted diffusion (vasogenic edema).50
Table 133-3 MRI Techniques and Traumatic Axonal Injury24,48,50,51
| MRI Technique | Features |
|---|---|
| Conventional Imaging | |
| T2 weighted | Limited in detecting periventricular/cortical lesions due to the presence of CSF nearby. |
| FLAIR | Very sensitive to nonhemorrhagic lesions. It suppresses the signal produced by CSF and detects a larger number of lesions than does T2-weighted MRI (ischemic TAI = hypersignal). |
| T2 weighted gradient echo | Very sensitive to paramagnetic blood breakdown products and thus to microhemorrhages (hemorrhagic TAI = hyposignal). |
| More Sensitive Sequences | |
| DWI | Detects alterations >3 hours after injury. DWI quantifies the overall restriction to water diffusion (ADC). There is an evolution from decreased ADC or restricted diffusion (cytotoxic edema) to increased ADC or unrestricted diffusion (vasogenic edema) in TAI lesions. |
| DTI and DTI tractography | Quantifies the directionality of water diffusion in three-dimensional space, providing in vivo information on white matter integrity. Water diffuses more freely in the direction of white matter fibers (FA). TAI is associated with FA reduction, which could be due to misalignment of fibers, edema, fiber disruption, or axonal degeneration. |
| Proton-MRS | Detects alterations since the injury. Proton-MRS assesses neurochemical alterations in the following metabolites: Detects alterations since the injury. Proton-MRS assesses neurochemical alterations in the following metabolites: • Cr, found in glia and neurons. Its level is believed to be stable and serves as a point of reference to calculate metabolic ratios. • NAA, an amino acid present in neurons. NAA reflects the status of neuronal tissue (lower NAA/Cr ratio in TAI). • Cho, component of cell membranes. Cho reflects glial proliferation or membrane breakdown (higher Cho/Cr ratio in TAI). • Lactate, a marker of anaerobic metabolism and ischemia (higher levels are seldom found in head-injured patients). |
ADC, apparent diffusion coefficient; Cho, choline; Cr, creatine; FA, fractional anisotropy; NAA, N-acetylaspartate, TAI, traumatic axonal injury.
T2-weighted, FLAIR, and gradient echo MRI sequences underestimate axonal injury. The novel technique of diffusion tensor imaging (DTI) has evolved in recent years as a valuable complementary tool to investigate DAI. DTI characterizes the directionality of water diffusion in a three-dimensional space, thus providing information on the physiologic status of particular axon bundles, a form of noninvasive in vivo tract tracing known as DTI fiber tractography (Fig 133-6 E).51 It evaluates the interruption of axolemmal transport and axonal leakage, being capable of detecting alterations 3 hours after injury. This technique has shown good correlation with the gold standard β-APP immunohistochemical method for detection of DAI in experimental models of TBI. Two pilot studies, one performed on 9 chronic TBI patients51 and the other on 12 head-injured patients within the first week after injury,52 showed post-traumatic damage in white matter tracts, such as the corpus callosum, fornix, and peduncular projections that were correlated with the outcome, and supported this technique as a promising diagnostic and prognostic tool (Fig 133-6 E).
Magnetic resonance spectroscopy (MRS) is a method used for assessment of neurochemical alterations since the moment of the injury. Proton-MRS is the commonly used method in vivo, in which a reduction in the N-acetylaspartate/creatine ratio and an increase in the choline/creatine ratio are the most characteristic detectable findings after TBI. These changes reflect, respectively, the degree of neuronal loss/membrane breakdown and the degree of glial proliferation.41 In addition, the MRS analysis of the biochemical profile in cerebral cortex and white matter of adult TBI patients studied 7 days after injury found that glutamate (Glu)/Glu+glutamine and Glu/choline ratios were significantly elevated in patients showing long-term outcomes of 4 or fewer points on the GOS 6 to 12 months after injury.53
Surgical Treatment of TBI
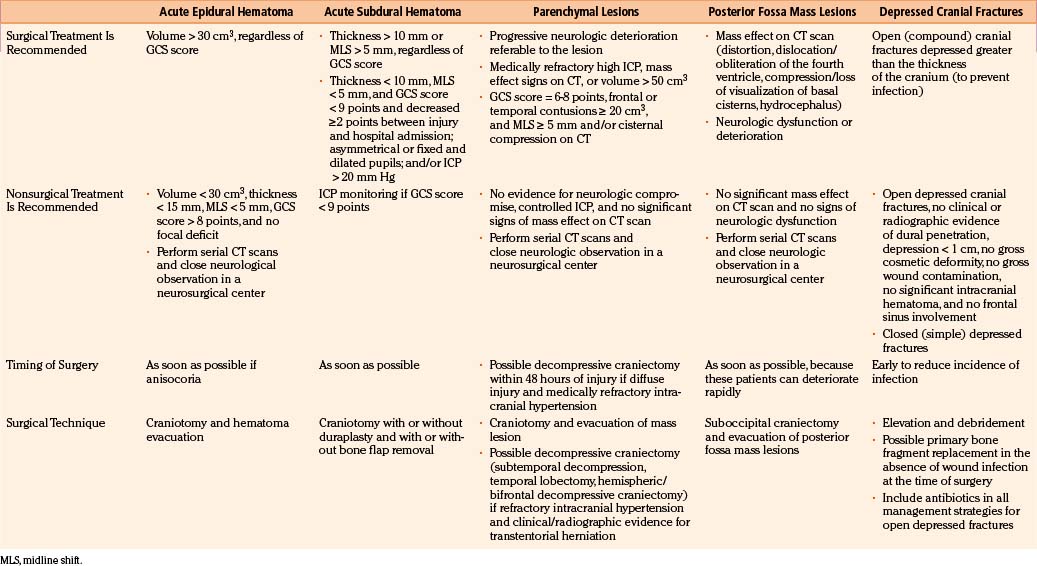
TBI treatment is an integrated team effort by surgeons and nonsurgeons. Probably the most important aspects of TBI care are nonoperative; however, the role played by neurosurgeons in the integral treatment of TBI patients remains critical, especially in making the appropriate decisions regarding whether to operate and, if operating, when to perform surgical procedures in advance to the progression of secondary damage.54 Surgical management of TBI has long been based on clinical anecdotal practice, assuming that any intracranial mass lesion in a patient with progressive neurologic deterioration should be evacuated as soon as possible by craniotomy. The new paradigm of evidence-based medicine has extend to the establishment of scientific guidelines for the surgical treatment of TBI, published in 2006, which should help neurosurgeons make the most appropriate decisions by using a strong scientific background, in addition to personal anecdotal experience.4,5 These guidelines were performed after a throughout analysis of the English-language TBI literature published since the use of CT scan. The appropriate articles were classified according to their scientific rigor in three classes of evidence with three associated levels of recommendation. No level I or II recommendation is provided by the surgical guidelines because of the lack of scientific class I or II evidence, mainly due to the ethical impossibility of carrying out prospective, randomized surgical trials. Consequently, guidelines can only formulate level III recommendations, reflecting unclear clinical certainty. As in all areas of clinical medicine, the optimal decision regarding treatment for an individual patient should not rigidly fall within a given recommendation of the guidelines but depend on a synthesis of the established knowledge based on guidelines and refined by the specific findings of the patient and the clinical judgment of the treating physician. A summary of the principal recommendations for surgical treatment of TBI lesions, together with the fundamental findings provided by the articles used for the elaboration of the guidelines, is provided in Table 133-4.
Surgical Treatment of Depressed Skull Fractures
Depressed skull fractures can be classified in two major types: (1) simple cranial depressed fractures, which have no galeal disruption and are only surgically managed if the extent of depression equals or exceeds the thickness of adjacent intact bone or if there is an associated intracranial hematoma with mass effect that requires evacuation, and (2) compound depressed cranial fractures, which have galeal disruption and an overlying scalp laceration in continuity with the fracture site and are generally treated with debridement and surgical elevation.4 Some compound depressed cranial fractures may be treated nonoperatively if there is no clinical or radiographic evidence of dural penetration, significant intracranial hematoma, depression greater than 1 cm, frontal sinus involvement, gross deformity, wound infection, pneumocephalus, or gross wound contamination.55 Several studies have also shown that the rate of postoperative infections is not increased by primary bone fragment replacement.56 There is no difference in the rate of postoperative infection between patients undergoing surgical repair of the fracture within 8 hours and those treated after that time. Anticonvulsants should be administered prophylactically to these patients, who are associated with a 20% or greater risk of developing seizures. Recommendations for the surgical treatment of depressed skull fractures, according to the surgical TBI guidelines, are listed in Table 133-4.
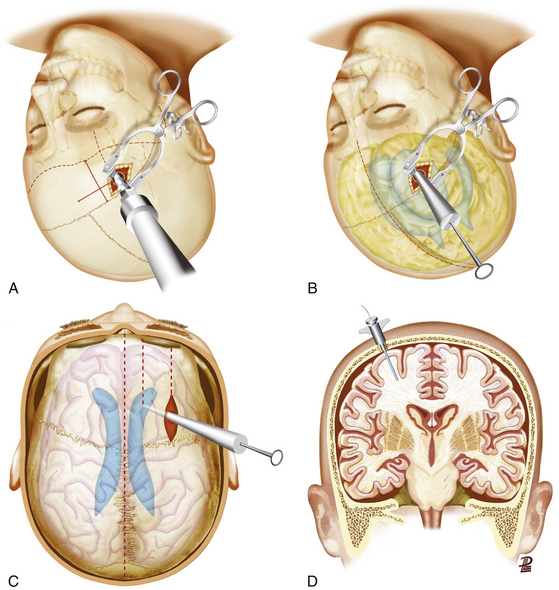
If the paranasal sinuses are involved by a depressed fracture, the goals of treatment are correction of the cosmetic deformity and prevention of a CSF fistula, regardless of whether the fracture is simple or compound. Depressed fractures of the anterior wall of the sinus do not result in a CSF fistula unless there is also a fracture of the posterior wall and a tear in the adjacent dura. A bicoronal scalp is fashioned, the pericranium is elevated as a separate layer, and the depressed fragments are elevated. Then the sinus mucosa is exenterated to prevent the formation of a mucocele, using a small drill if necessary. Next, the residual cavity can be filled with temporalis muscle and fat (Fig. 133-6 C). A pericranial graft can then be reflected over the sinus and sewn to the dura to further isolate it from the intracranial contents. The bone fragments comprising the anterior wall of the sinus are wired together and secured to the skull. The bone fragments of the posterior wall do not need to be replaced.
Post-Traumatic Intracranial Hematomas: Surgical Versus Nonoperative Management
The formation of intracranial hematomas after a head injury is a common occurrence. Among the 746 severely head-injured victims included in the TCDB, 37% of patients underwent surgery for removal of an EDH, SDH, or intracerebral hematoma (ICH).57 To detect hematomas at an early stage, it is essential to identify the risk factors that increase the changes that a given patient will be harboring an intracranial hematoma. They consist of the presence of a skull fracture on radiograph, any neurologic dysfunction, a seizure or severe headaches and vomiting, and a decrease in the level of consciousness. Patients with blood coagulation disorders should be observed particularly carefully for progressive enlargement of hematomas, and the clotting disorders must be vigorously treated. Overall, nonoperative management of SDHs and EDHs should be considered only if the patient is fully conscious, the extra-axial mass lesion is the single dominant lesion (i.e., there are no multiple contusions or potentially significant contralateral mass lesions, which may be preventing midline shift), and there are no features of mass effect, such as midline shift greater than 3 mm, or basal cistern effacement. Finally, there are situations in which the dismal clinical status of the patient warrants a nonoperative course despite the presence of a radiographic surgical lesion. Such cases include the adult patient who is moribund and flaccid with a GCS score of 3 points, nonreactive and dilated pupils, and no spontaneous respirations.58 Similarly, in patients older than 75 years of age with a GCS score of 5 or fewer points, a nonoperative course usually should be taken, given their consistently poor outcome with or without surgery.
High ICP and ICP Monitoring in TBI
The brain is housed in a rigid cavity and has a limited ability to compensate any volume occupation caused by intracranial hemorrhages or the development of swelling. Clinical examinations are unreliable unless the patient has progressed to brain stem herniation. The most accurate way of assessing ICP is to insert a monitor to allow the clinician to anticipate irreversible brain damage by a prompt medical or surgical intervention. There is substantial evidence that ICP monitoring may improve outcome, because it helps in earlier detection of intracranial mass lesions and guides therapies to control intracranial hypertension. About two thirds of patients with severe head injury have no significant mass lesion on the initial CT scan, but at least 80% demonstrate high ICP.59 It is generally accepted that severely head-injured patients (GCS score 3-8 points) with abnormal scans should undergo ICP monitoring. Effaced cisterns are associated with high ICP in more than 70% of cases, and mandatory ICP monitoring is advocated in such cases. The prevalence of postoperative intracranial hypertension is extremely high (greater than 75%) in patients who were in a coma before removal of an intracranial hematoma. In such patients, therefore, an ICP monitoring device should be inserted at the end of the procedure.
The gold standard method for measuring ICP is an intraventricular device connected to a fluid-coupled catheter with an external strain gauge (ventriculostomy catheter), because it not only monitors ICP but also provides CSF drainage to lower ICP (Fig. 133-7A-C). Nevertheless, many centers tend to use intraparenchymal probes, especially when the ventricular size is small and it is expected to need a high number of passes. This device should be inserted through a bur hole and skin incision separated from the craniotomy. A twist drill is usually made just anterior to the coronal suture in line with the pupil. Following the opening of the dura with a blunt stylet, the transducer tip is directly placed in the brain parenchyma at a distance of 2 to 3 cm into the brain parenchyma (Fig. 133-7D).
Surgical Timing in Post-Traumatic Lesions
In general, the decision regarding whether or not to proceed with surgical evacuation of a post-traumatic hematoma depends on the patient’s clinical condition, neurologic status, and imaging findings. Clinical features indicating high ICP or any impairment to the level of consciousness, oculomotor paresis ipsilateral to a sizable hematoma, bradycardia, severe headache, repeated vomiting, and/or arterial hypertension. CT features suggesting raised ICP are shift of brain structures, obliteration of all subarachnoid CSF spaces, dilatation of the lateral ventricle contralateral to the hematoma, and hematomas of large size. A prompt surgical evacuation is essential for masses producing brain stem compression because of the risk of irreversible ischemic brain damage. In these cases, the sooner the decompression is achieved, the greater the possibility of good recovery. There is clear evidence that delayed evacuation of an EDH/SDH causing significant midline shift once clinical deterioration is present is associated with much poorer results, especially if the evacuation is later than 2 to 4 hours of the onset of the coma.60
Greater controversy exists over when to evacuate parenchymal brain contusions and traumatic ICHs and whether such removal is helpful in controlling ICP and improving outcome.61 Miller et al., in their experience with more than 200 severely head-injured patients, contusions, and traumatic hematomas associated with the highest incidence of poor ICP control despite their surgical removal, recommended an initial nonoperative management for such lesions.61 Other authors believe that early surgical removal of larger hematomas and cerebral contusions provides early control of ICP and helps prevent the cascade of secondary damage. Temporal lobe hematomas are especially likely to cause brain stem compression at low ICP with little midline shift and should be evacuated sooner than other locations. An initial conservative approach is warranted, however, when eloquent cortex is involved. Similarly, intracranial hematomas confined to deep white matter or the basal ganglia are best managed without surgery.
Acute EDH
An acute EDH develops between the dura mater and the inner table of the skull and is reported in 2.5% to 5% of head-injured patients (Fig. 133-4A). EDHs adopt a lens-shaped or biconvex morphology as the periosteal layer of dura is stripped from the inner table of the skull by the bleeding. The classic and most lethal type of EDH is caused by a skull fracture crossing the groove in the temporal bone containing the middle meningeal artery. These hematomas generally accumulate rapidly, although a delayed stepwise progression over hours may also occur. EDHs may have also a venous origin, either from diploic veins or from bridging veins torn along their course into venous sinuses. These lower pressure hematomas develop slowly and are often self-limited, but no reliable imaging signs can be used to assist in their prediction, and this subtype should be considered dangerous. Some EDHs accumulate from rebleeding, accounting for up to 30% of all EDHs in some series.62 Dural stripping by the blow itself has been postulated to be the alternative presumed mechanism. Epidural collections arising solely from skull fracture bleeding are most often small and most frequently do not expand in the absence of damage to an adjacent sinus or coagulopathy. Nevertheless, nonoperative management based on anticipated stability should be supported by serial imaging.
The “classic” lucid interval consisting of a brief LOC caused by the blow per se followed by an awaken period and late deterioration due to rising ICP and mass effect (patients who “talk and die”) only occur in a minority of cases, approximately 15% to 30%.63 Between 20% and 55% of the patients are comatose on admission or immediately before surgery.62,64 Although most EDHs are diagnosed on the initial CT scan, the clinician must remain alert to the possibility of an EDH that develops or rapidly enlarges after an apparently negative scan. A vertex hematoma may be missed on a routine axial CT scan, but a coronal reconstruction should identify the lesion.
EDHs are not usually associated with underlying parenchymal damage, so their early surgical evacuation is highly successful. Global mortality of a surgically treated EDH is approximately 10%.62,64 The surgery is usually an emergency, especially in patients in a coma at the time of the diagnosis, as the duration from time of injury to treatment is an important determinant of prognosis. Other factors in determining the outcome are age, depth of coma, degree of midline shift, and size of hematoma. The mortality rate and outcome after evacuation of an EDH is directly related to the level of consciousness before surgery, ranging from 0% for patients conscious at the time of operation to 27% for patients presenting with the classic lucid interval and reaching 50% for patients who never regain consciousness after the impact.
In addition, neurosurgeons should bear in mind the possibility of a delayed EDH, which accumulates after an initial normal CT scan. These may occur in 4% to 30% of EDHs series and be associated with a slightly higher morbidity and mortality because many patients developing such lesions are diagnosed and operated on after the apparition of neurologic deterioration.62 Mechanisms involved in their origin include correction of hypotension or hypovolemia, pseudoaneurysm, or coagulopathy.
Management
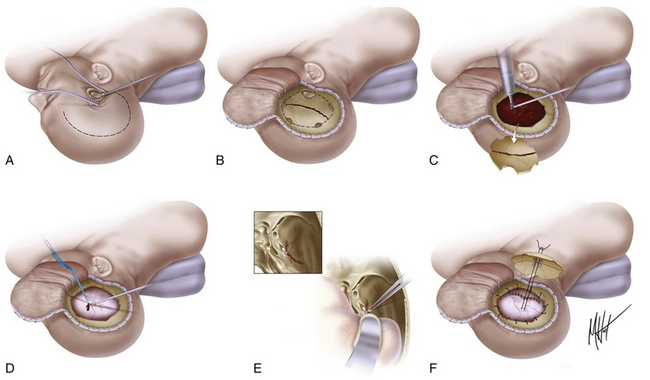
There are no class I or II evidence-based guidelines for the specific management of EDHs, and treatment is based on the patient’s GCS score, pupillary exam, comorbidities, CT findings, and age and the surgeon’s personal experience.4 An expanding temporal EDH in a deteriorating patient is a surgical emergency, whereas a small frontal EDH in an awake, alert, and talking patient can usually be managed conservatively. Chen et al. analyzed factors associated with delayed neurologic deterioration and the need for surgical evacuation in initially nonoperated cases and found that volumes greater than 30 ml, clot thickness greater than 15 mm, or midline shift greater than 5 mm on the initial CT scan was statistically associated with failed indication of conservative treatment.65 When clinical observation and serial imaging are the chosen procedure, the patient must be admitted to an intensive care unit and the first follow-up CT scan should be obtained within 6 to 8 hours after TBI. No enlargement of an EDH has been observed later than 36 hours. For the recommended surgical options versus conservative treatment, surgical techniques, timing, and outcome, refer to Table 133-4 and Fig. 133-8.
< div class='tao-gold-member'>
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Tumors Involving the Cavernous Sinus
Tumors Involving the Cavernous Sinus
 Disc Replacement Technologies in the Cervical and Lumbar Spine
Disc Replacement Technologies in the Cervical and Lumbar Spine
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


