Chapter 44 Surgical Management of Tumors of the Jugular Foramen
Historical Background
In 1840, Valentin1 observed a small cellular formation near the origin of the tympanic nerve that he identified as ganglionic tissue and termed “ganglionium tympanicum” or “intumescentia gangliosa.” In 1878, Krause2 further demonstrated highly vascular glomus tissue along the tympanic branch of the glossopharyngeal nerve (“glandula tympanica”) that was histologically similar to the carotid body, as reported by Von Lushka in 1862.3 This work received little attention until 1941, when Guild4,5 described glomus tissue as a flattened ovoid body in the adventitia of the dome of the jugular bulb and coined the term “glomus jugularis” to describe paraganglionic tissue composed of capillary or precapillary vessels interspersed with numerous epithelioid cells found along the jugular bulb. In sectioning human temporal bones, Guild found that 50% of this tissue occurred in the jugular bulb, approximately 25% occurred along the course of the tympanic branch of the glossopharyngeal nerve (Jacobson’s nerve), and 25% occurred along the auricular branch of vagus nerve (Arnold’s nerve). This observation explained the existence of “glomus tumors” that occurred both in the middle ear (glomus tympanicum tumors) and in the region of the jugular bulb (glomus jugulare tumors). Rosenwasser6 in 1952, was the first to suggest a possible relationship between the “glomus jugularis” and the “carotid body–like” tumors in the temporal bone. The designation “tumors of the glomus jugulare” was first mentioned by Lattes and Waltner in 1949.7
Management of jugular foramen tumors has evolved over the years. The inaccessibility of the jugular foramen because of its relatively deep location, high vascularity, and proximity to cranial nerves made tumor management extremely challenging; surgery in this region was often associated with a poor outcome. Surgery in the 1930s was primarily performed using a suboccipital approach with removal of bone around the jugular foramen to avoid excessive bleeding.8 A subtotal resection followed by radiation therapy was generally performed.9–12 Most patients had resultant postoperative lower cranial palsies. Mobilization of the facial nerve to better access the jugular foramen was first described by Capps13,14 in 1952 and later by others.15,16 Capps combined this with proximal and distal control of the sigmoid sinus and the jugular vein; however, attempts to remove the jugular bulb were met with excessive bleeding and poor outcomes.
In the 1960s and 1970s, the advent of better surgical technology resulted in better surgical outcomes. These innovations included the operating microscope, microneurosurgery dissection techniques, bipolar electrocautery, safer neuroanesthesia, arteriography,17,18 polytomography,19 retrograde jugular venography,16,20 computed tomography (CT),21 and magnetic resonance imaging (MRI).22 Hearing preservation surgeries were advocated by House and Glasscock23 and Farrior24 (modified endaural postauricular hypotympanotomy). In 1969, McCabe and Fletcher25 proposed that the size and extent of the tumor were the determining factors for selecting the most appropriate surgical approach. Soon after, new classification schemes were proposed by Fisch26 and by Jackson et al.27 based on tumor size, intracranial extension, and surgical operability. The 1970s saw the emergence of multidisciplinary skull base approaches,27 including the combined lateral skull base approach (suboccipital craniectomy with mastoidectomy),28–30 and infratemporal fossa approaches by Fisch31 (modified after Farrior’s hypotympanic approach). Other modifications to access the jugular foramen using skull base approaches were further advocated by Al-Mefty and colleagues,32–34 Bordi et al.,35 Patel et al.,36 and Liu et al.37
Despite the improvements in surgical exposure, extreme vascularity of the tumor was still a major challenge during surgery. The introduction of preoperative superselective transarterial embolization38–41 of jugular foramen tumors significantly reduced the vascularity of the tumor, making surgery safer.
Anatomic Considerations of the Jugular Foramen
The jugular foramen, also called the posterior foramen lacerum, is situated in the posterior fossa lateral to the carotid canal. The walls of the jugular foramen are formed anterolaterally by the petrous bone and posteromedially by the occipital bone.42,43 The foramen is directed in an anterior, lateral, and inferior direction. According to morphometric studies, the jugular foramen can be more accurately described as a triangular canal with an endocranial (~14.5 × 7 mm) and an exocranial opening (~9 × 17 mm). It lies about 23 mm medial to the apex of the mastoid tip, 15 mm medial to the tympanomastoid suture, and 5 mm above the intracranial orifice of the hypoglossal canal.44–47
The pars venosa (or pars vascularis) is situated in the posterolateral aspect of the jugular foramen and contains the internal jugular vein (IJV), the jugular bulb, the posterior meningeal branch of the ascending pharyngeal artery, the vagus nerve (cranial nerve X), the auricular branch of the vagus nerve (Arnold’s nerve), and the spinal accessory nerve (cranial nerve XI). A smaller pars nervosa is located in the anteromedial portion of the jugular foramen and contains the glossopharyngeal nerve, the tympanic branch of the glossopharyngeal nerve (Jacobsen’s nerve), and the inferior petrosal sinus. This nomenclature can be somewhat misleading, because both structures contain neural, as well as vascular, structures.44
The important structures surrounding the jugular foramen include the mastoid segment of the facial nerve laterally, the petrous segment of the internal carotid artery (ICA) anteromedially, the vertebral artery inferiorly, and the hypoglossal nerve medially. Access to the jugular foramen from a lateral trajectory is obstructed by the mastoid and styloid processes, the transverse process of C1, and the mandibular ramus.42 The deep potential spaces along the foramen include the middle layer of the deep cervical fascia (buccopharyngeal fascia) anteromedially, the deep layer of the deep cervical fascia (prevertebral fascia) posterolaterally, and the superficial layer of the deep cervical fascia laterally.44 These potential spaces are important in understanding the spread of tumors in this region.
The cranial nerves run anteromedially to the jugular bulb and maintain a multifascicular histoarchitecture, especially cranial nerve X, which is formed by multiple fascicles, in contrast to cranial nerves IX and XI, which are formed of only one or two fascicles. The tympanic branch of the glossopharyngeal nerve (Jacobson’s nerve) and the auricular branch of the vagus nerve (Arnold’s nerve) cross the jugular foramen. A dural septum separates cranial nerve IX from the fascicles of cranial nerves X and XI.45,46,48 The only intradural site at which the glossopharyngeal nerve is consistently distinguishable from the vagus nerve is just proximal to this dural septum. In about 23% to 30% cases,44 a bony partition of the jugular foramen may be seen. The facial nerve exits the stylomastoid foramen approximately 5 mm lateral to the lateral edge of the jugular foramen. The hypoglossal nerve does not traverse the jugular foramen; however, it joins the nerves exiting the jugular foramen just below the skull base and runs with them in the carotid sheath.42 Tumors in this area can cause Vernet syndrome (jugular foramen syndrome),49 which is characterized by paralysis of cranial nerves IX, X, and XI caused by tumor expansion within the jugular foramen.
The muscular relationships encountered during surgery at the jugular foramen include the sternocleidomastoid (SCM), situated superficially in the lateral neck, and the splenius capitis, longissimus capitis, levator scapulae, and scalenus medius muscles in a deeper muscular layer. Anteriorly, the posterior belly of the digastric muscle is encountered. Immediately laterally, the styloid process and its attached musculature (stylopharyngeus, stylohyoid, and styloglossus muscles) appear in a triangular zone bounded by the posterior belly of digastrics muscle, the external auditory canal, and the mandibular ramus. Displacement of the digastric muscles exposes the transverse process of C1 (where the superior and inferior oblique muscles attach). The rectus capitis lateralis is the muscle most intimately related to the jugular formen.42
Specific Pathologic Conditions
Glomus Jugulare Tumors
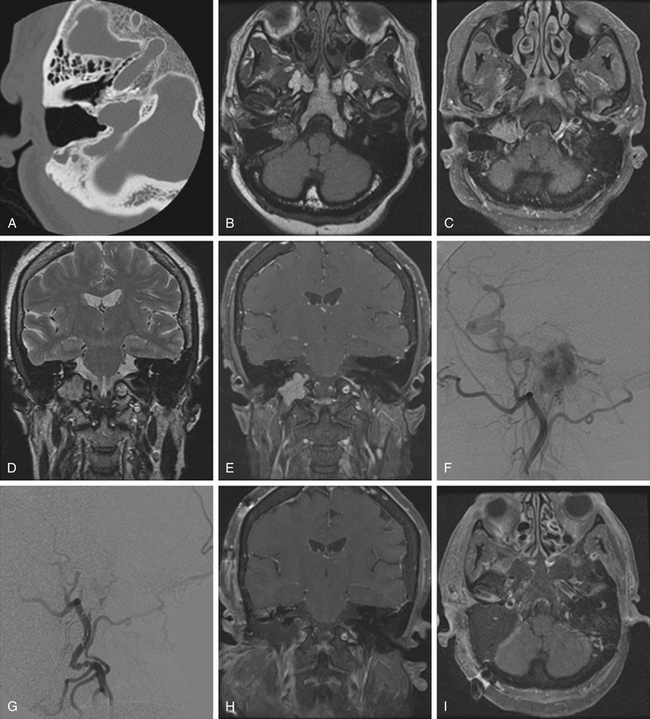
Glomus jugulare tumors are rare lesions with an annual incidence of approximately 1 in 1.3 million people per year.50,51 Women are affected more often than men (6:1 ratio).52,53 Glomus tumors have also been referred to as paragangliomas, chemodectomas, nonchromaffin tumors, glomerocytomas, and receptomas. Glomus jugulare tumors arise from paraganglia in the adventitia of the jugular vein. In contrast, glomus tympanicum tumors arise from paraganglia associated with Arnold’s and Jacobson’s nerves within the middle ear. Tumors occur most frequently in patients between 40 and 70 years of age, but some have been reported in patients as young as 6 months and as old as 88 years. Although they are histologically benign and only rarely metastatize54 or display malignant features,55 these tumors may locally invade neighboring structures, such as bone, dura, carotid artery, and cranial nerves.48 Jansen et al.56 estimated the doubling time of the size of head and neck paragangliomas to be 13.8 years, with an annual growth rate of 0.79 mm/yr. The incidence of multicentric glomus tumors is 3.7% to 10% in sporadic cases57,58 and up to 55% in familial cases.59 For familial glomus tumors, the mean age of patients at presentation is younger, almost 50% of patients have multicentric tumors, and a higher proportion have bilateral tumors.60,61 Four hereditary paraganglioma syndromes have been described, referred as PGL 1 to 4.62
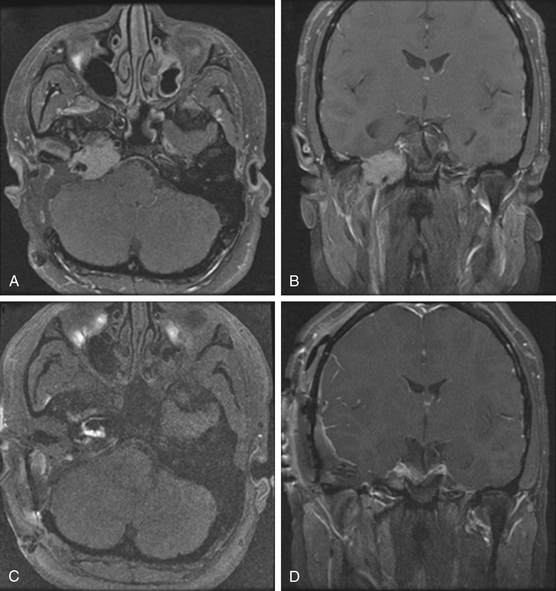
Pathologically, glomus tumors appear as highly vascular nonencapsulated masses (Figs. 44-1 and 44-2). Histologically, they are indistinguishable from glomus tympanicum, glomus vagale, and carotid body tumors. These tumors are characterized by large groups of polyhedral epithelioid cells (chief cells) with centrally located uniformly hyperchromatic nuclei and finely granular cytoplasm.63 There is an extensive thin-walled capillary and reticulin network surrounding the nests of chief cells, creating the characteristic Zellballen appearance.48 Immunohistochemical analysis of chief cells demonstrates positivity for chromogranin, synaptophysin, neuron-specific enolase, and neurofilament. Malignant glomus tumors demonstrate immunoreactivity to MIB-1, p53, Bcl-2, and CD34. The extreme vascularity of these tumors is due to angiogenesis from vascular endothelial growth factor and platelet-derived endothelial cell growth factor.64 Ultrastructurally, secretory granules containing norepinephrine, epinephrine, and dopamine may be seen. Because of the presence of catecholamines and neuropeptides, they are included in the amine precursor uptake and decarboxylase system65 or the diffuse neuroendocrine system.
Schwannomas
Schwannomas are the second largest group of tumors at the jugular foramen (Figs. 44-3 to 44-5).66 Ninety percent of jugular foramen schwannomas originate from cranial nerve IX or X.67,68 Samii et al.69 classified these tumors into four subtypes based on location. Type A tumors are those that primarily occupy the cerebellopontine angle with minimal enlargement of the jugular foramen. Type B tumors are primarily located in the jugular foramen with intracranial extension. Type C consists of primarily extracranial tumors with extension into the jugular foramen. Type D consists of dumbbell-shaped tumors with both intra- and extracranial components.
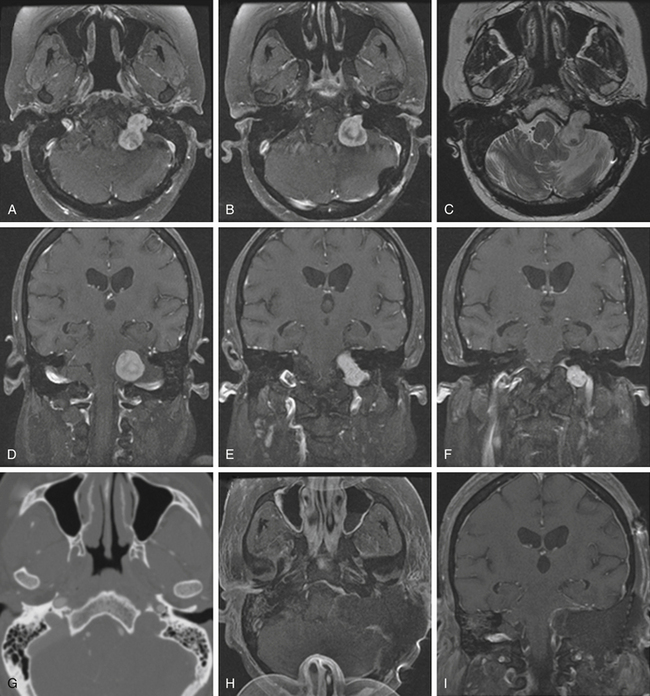
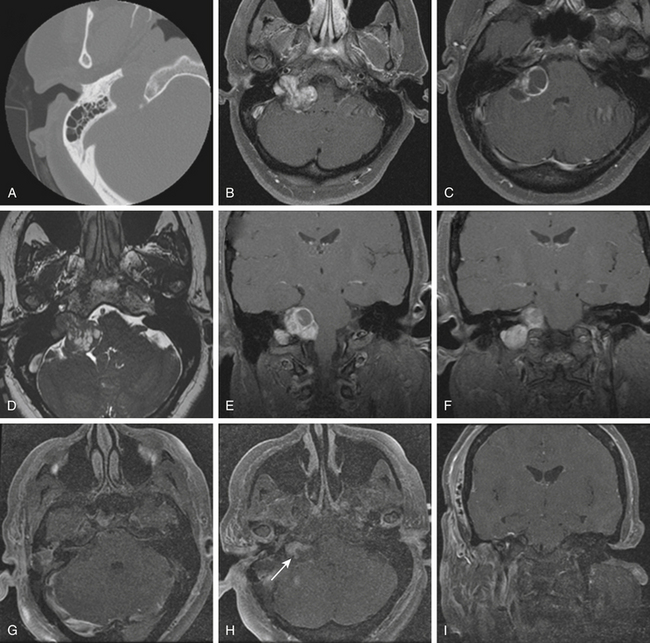
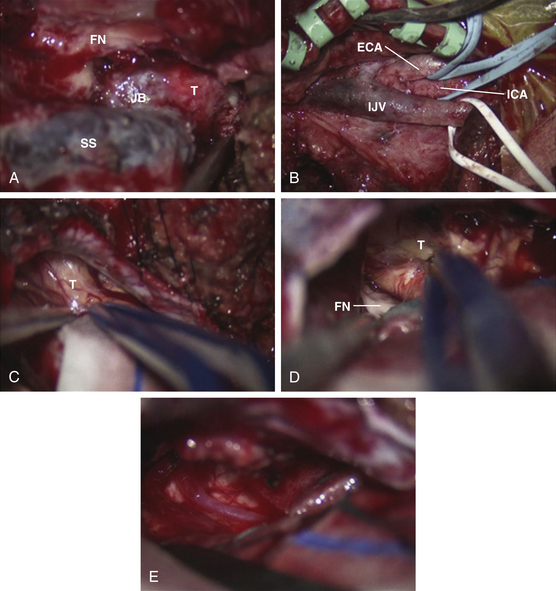
On unenhanced CT scans,70 schwannomas appear isodense to the brain and enhance brightly as well-demarcated tumors after contrast administration. As opposed to glomus tumors, the bony remodeling of the jugular foramen appears smoothly scalloped and well corticated. On T1-weighted MRI,71 schwannomas are isointense to brain and markedly enhance after gadolinium administration. On T2-weighted imaging, they are hyperintense to brain. On angiography, schwannomas do not appear very vascular and often compress the jugular bulb, in contrast to glomus tumors, which are highly vascular and invade the jugular bulb.71
At surgery, jugular foramen schwannomas appear as well-circumscribed, firm or rubbery, tan–white masses. Histologically, they comprise alternating patterns of compact spindle cells called Antoni A areas, interspersed with loose, hypocellular regions called Antoni B areas. Whorling or palisading formation of the nuclei may be seen. The hyperchromatic nucleus is elongated and twisted with indistinct cytoplasmic borders. These tumors often demonstrate retrogressive changes in the form of cystic degeneration, hyalinization, necrosis, calcification, and hemorrhage. Immunohistochemically, they demonstrate a uniformly intense S-100 protein positivity.72 Malignant transformation of these benign lesions is rarely seen.49
Meningiomas
Although basal posterior fossa meningiomas often extend into the jugular fossa, primary jugular foramen meningiomas are rare, with fewer than 100 cases reported in the literature.73–76 The jugular foramen as a location for primary meningiomas accounts for only 4% of all posterior fossa meningiomas.77 Primary jugular foramen meningiomas presumably arise from the arachnoid cap lining the jugular bulb and are more common in women (2:1).78 These meningiomas tend to exhibit an invasive growth pattern with extensive skull base infiltration, differentiating them from the meningiomas that are centered in the posterior fossa and secondarily extend into the jugular foramen.44,79 These lesions infiltrate surrounding temporal bone and neurovascular structures and require wide margins of excision to minimize risk of recurrence. A centrifugal pattern of spread, a permeative-sclerotic appearance of the bone margins of the jugular foramen, the presence of dural tails, and an absence of flow voids are particularly important features that assist in differentiating these from more common jugular foramen tumors. On CT scans, they appear isodense to the brain and show marked enhancement with contrast administration; there is occasional calcification and infiltration of the skull base with a characteristic hyperostosis.75
At surgery, they are solid, well-circumscribed extra-axial masses and are usually sessile with a broad dural base. On MRI studies, they are typically isointense to hypointense on T1-weighted imaging and enhance markedly after gadolinium administration. Moreover, the angiographic blush time is significantly longer than that of a glomus tumor. As compared with surgical intervention for glomus tumors or schwannomas, surgery on primary jugular foramen meningiomas often has a worse cranial nerve outcome76 (approximately 60% for meningiomas, 30% for glomus jugulare tumors, and 15% for schwannomas).80 There is also a higher recurrence rate of up to 25% at 5 years after resection.79
Classification
The main pathways of direct tumor spread are important in understanding the clinical and neurologic manifestations, operative strategies, and risk assessment. Glomus tumors tend to expand within the temporal bone via the pathways of least resistance, that is, air cells, vascular lumens, skull base foramen, and the Eustachian tube.81,82 According to Spector et al.,83 glomus tumors can spread through the following pathways once they have locally invaded into the temporal bone and middle ear: (1) down the Eustachian tube into the nasopharynx and then through the skull base foramina, (2) along the carotid artery into the middle fossa, (3) along the jugular vein or hypoglossal canal toward the posterior fossa, (4) through the tegmen tympani to the middle fossa floor, or (5) through the round window, with extension via the internal auditory canal into the cerebellopontine angle.83,84 Extension within the sigmoid and inferior petrosal sinus may be present as well. Although glomus tumors are usually considered benign and locally invasive, distant metastases to cervical lymph nodes, lung, and bone have been reported in about 4% to 19% of cases.84,85
The two most commonly used classifications of glomus tumors are those proposed by Fisch and colleagues26,86 and Jackson et al.27 (Tables 44-1 and 44-2). These systems are based primarily on tumor location and size. In 2002, Al-Mefty and Teixeira32 defined a subgroup of complex glomus jugulare tumors as those having one or more of the following criteria: giant size, multiple paragangliomas (bilateral or ipsilateral), malignancy, catecholamine secretion, association with other lesions such as dural arteriovenous malformations or adrenal tumors, or previous treatment with adverse outcome (prior surgery, radiation, or embolization).
TABLE 44-1 Fisch Classification
| Fisch Grade | Extent of Tumor |
|---|---|
| A | Middle ear cleft (glomus tympanicum) |
| B | Tympanomastoid area with no infralabyrinthine compartment involvement |
| C | Infralabyrinthine compartment of the temporal bone and extending into the petrous apex |
| C1 | Limited involvement of the vertical portion of the carotid canal |
| C2 | Invasion of the vertical portion of the carotid canal |
| C3 | Invasion of the horizontal portion of the carotid canal |
| D1 | Intracranial extension <2 cm in diameter |
| D2 | Intracranial extension >2 cm in diameter |
TABLE 44-2 Glasscock-Jackson Classification
| Glasscock-Jackson Grade | Extent of Tumor |
|---|---|
| I | Involves the jugular bulb, middle ear, and mastoid; generally small in size |
| II | Extends under the internal auditory canal; intracranial canal extension possible |
| III | Extends into the petrous apex; intracranial canal extension possible |
| IV | Extends beyond the petrous apex into the clivus or intratemporal fossa; intracranial canal extension possible |
Clinical Presentation and Diagnosis
Patients with jugular foramen tumors often present with initial symptoms of hearing loss and swallowing difficulty, as well as pulsatile tinnitus with glomus tumors. Invasion of the middle ear results in conductive hearing loss. The glomus tumor may erode through the floor of the hypotympanum, presenting as a middle ear mass; if there is erosion of the tympanic membrane, it may present as an aural polyp. Extension of the tumor through the facial recess results in facial nerve encasement and a deficit of the facial nerve.87,88
Glomus tumors have been associated with multiple endocrine neoplasia type 2, neurofibromatosis type 1, and Von Hippel–Lindau syndrome.87 Familial paragangliomas, in contrast to sporadic cases, are commonly multiple and bilateral, present at an earlier age, and are inherited almost exclusively via the paternal allele.53,89 Genetic mapping has identified loci 11q13 and q23 as sites likely responsible for tumorogenesis.90
Although the incidence of catecholamine secretion is approximately 4%,52,91 complications of blood pressure and pulse fluctuations intraoperatively can occur during surgical manipulation.32 Therefore, preoperative assessment for catecholamine secretion should be performed, including urinary vanillylmandelic acid and metanephrines.92,93 An abdominal CT or MRI should be obtained in all such patients to rule out a pheochromocytoma as an adrenal tumor source of catecholamine secretion.32 In patients with catecholamine-secreting tumors, pretreatment with alpha- or beta-blockers is warranted. Epinephrine release during surgical manipulation can provoke a hypertensive crisis, whereas histamine and bradykinin release can cause severe hypotension.94
Radiologic Imaging
On thin-section axial CT, glomus jugulare tumors produce a characteristic “moth-eaten” pattern of destruction of the temporal bone and adjacent structures, particularly the jugular spine and the carotid crest (the bone separating the petrous carotid artery from the jugular bulb). These tumors can also cause dehiscence of the floor of the tympanic cavity, with extension into the middle ear and destruction of the bony labyrinth. The tendency of glomus jugulare tumors to erode bone helps distinguish them from glomus tympanicum tumors, which are generally smaller and arise from the cochlear promontory, enveloping but not usually destroying the ossicular chain.44 In some cases, however, the differentiation cannot be made between the two tumors; therefore, they are called glomus jugulotympanicum tumors. On T1-weighted MRI, glomus tumors demonstrate a characteristic “salt and pepper” pattern caused by flow voids within the highly vascular tumor.95 They enhance heterogeneously after gadolinium administration.96 More recently, 111indium octreotide, a radiologic somatostatin analogue, has been used to selectively identify paragangliomas, especially for detecting multicentricity, recurrence, and metastatic disease.97
Jugular foramen schwannomas, on the other hand, enlarge the jugular foramen with widened smooth, scalloped, sclerotic margins seen on CT scans. Unlike meningiomas, there is a lack bony invasion in schwannomas.95,98 Meningiomas frequently invade bone, particularly the jugular spine and jugular tubercle, resulting in hyperostosis.96 Schwannomas are usually solid and well circumscribed but occasionally demonstrate cystic degeneration. On MRI, they appear hypointense on T1-weighted images and hyperintense on T2-weighted images; they also brightly enhance, though the enhancement can be heterogeneous if there is associated necrosis or cystic degeneration.95 When there are intra- and extracranial components, a pathognomonic dumbbell-shaped tumor can be seen best on coronal or sagittal views. Meningiomas typically appear isointense to hypointense on T1-weighted images and enhance considerably after gadolinium administration. A pathognomonic “dural tail” is usually seen.
On diagnostic cerebral angiography, glomus jugulare tumors demonstrate an intense tumor blush because of their hypervascularity.95 Large feeding vessels and early draining veins are commonly associated with glomus tumors. The main blood supply of glomus tumors comes from the external carotid artery via the inferior tympanic branch of the ascending pharyngeal artery.89 Large tumors may also parasitize their blood supply from meningeal branches of the occipital artery, the posterior auricular artery, caroticotympanic branches from the petrous ICA, ascending cervical branches from the thyrocervical trunk, the posterior inferior cerebellar artery, and the vertebral artery.87,96 Narrowing and irregularities of the ICA may indicate tumor infiltration of the carotid wall. In comparison with glomus tumors, meningiomas and schwannomas are only mild to moderately vascular. Meningiomas of the jugular foramen do not necessarily demonstrate the early, prominent, and prolonged tumor blush that is frequently seen with supratentorial meningiomas.96 Cerebral angiography is also useful in demonstrating the patency of the sigmoid sinus and jugular vein to assess the venous outflow of the tumor during preoperative planning.
Related posts:
 Cortical and Subcortical Brain Mapping
Cortical and Subcortical Brain Mapping
 Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
Radiation Therapy and Radiosurgery in the Management of Craniopharyngiomas
 Surgical Management of Intracerebral Hemorrhage
Surgical Management of Intracerebral Hemorrhage
 Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
Neurovascular Decompression in Cranial Nerves V, VII, IX, and X
 Tumors Involving the Cavernous Sinus
Tumors Involving the Cavernous Sinus
 Arachnoid, Suprasellar, and Rathke’s Cleft Cysts
Arachnoid, Suprasellar, and Rathke’s Cleft Cysts



