and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
The most common nerve biopsy findings are nonspecific axonal alterations such as axonal degeneration, depletion, and regeneration. The pathologist can assess the severity and chronicity of the damage, but infrequently can find an unequivocal structural indication of its underlying cause. Two major patterns of axonal disease can be identified: those changes indicating a disturbance of axonal metabolism (axonopathy) and those changes indicating axonal degeneration (Wallerian degeneration). Although electron microscopy provides a more detailed view of normal and pathological features than light microscopy, the stereotyped changes of Wallerian degeneration or axonopathy represent one final common pathway of disease and do not permit specific diagnosis of the underlying etiology. Only rarely do axonal alterations have specific implications, for example, giant axonal swellings with filamentous accumulations or the classic features of neuroaxonal dystrophy.
We shall review the normal ultrastructure of axons; certain aspects of axonal physiology necessary to facilitate understanding of pathogenic mechanisms throughout this book; and the two main variants of axonal injury, Wallerian degeneration, and distal axonopathy. The chapter concludes with a discussion of axonal regeneration.
4.1 The Normal Axon
The axon is a cell process specialized for the conduction of electrical impulses to or from the cell body. While other important functions exist, such as bringing back to the cell body (perikaryon) information regarding the biochemical and hormonal milieu of the nerve terminal, the rapid linear propagation of electrical impulses is the feature that makes the axon unique. The cell bodies of cutaneous sensory axons examined by nerve biopsy lie in spinal ganglia with the axonal diameter roughly proportional to cell body size. Because of the great length of peripheral nerve fibers, the volume of cytoplasm in the axon exceeds that of the perikaryon by several orders of magnitude.
4.1.1 Contents of the Normal Axon
Although recent studies have shown the existence of scattered ribosomes in axons which are likely to have an important role in normal axonal function (Koenig et al. 2000) and in pathological processes (Court et al. 2008), the most essential macromolecules must be synthesized in the cell body and transported centrifugally. An elaborate architecture and physiology have evolved in order to carry out this process (recently reviewed in Brown 2013). The normal axon contains microtubules, neurofilaments, mitochondria, membrane-bound tubulovesicles, and various dense bodies and granules (Fig. 4.1a, b). Many of these organelles are involved in the axonal transport process.
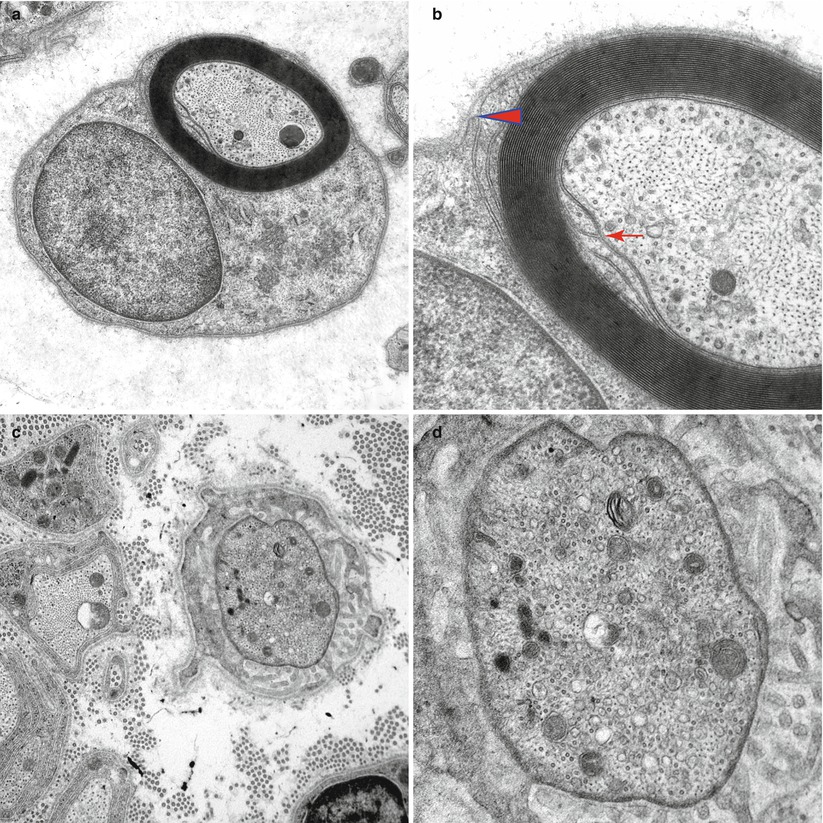
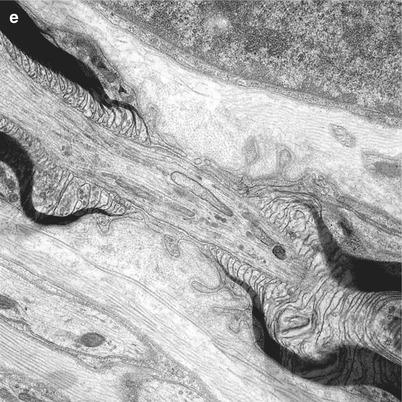
Fig. 4.1
Normal axon. Transverse section at internode (a, b) showing the internal (arrow, b) and external (arrowhead, b) mesaxons with central neurofilaments and peripheral microtubules. (c, d) Node of Ranvier with increased organelle density. (e) Longitudinal section through the node of Ranvier (Magnification, a 20,000×; b 50,000×; c 20,000; d 50,000×; e 12,000×)
4.1.1.1 Neurofilaments and Microtubules: The Cytoskeleton
Classified as intermediate filaments, neurofilaments measure 10 nm in diameter. These structures, which are the most abundant axoplasmic organelles, align parallel to the axon and are evenly distributed with a density of about 100–300 filaments per μm2 in cross sections of myelinated and unmyelinated axons of various species (Berthold 1978). In myelinated fibers of cat (Table 2 in Berthold 1978) and mouse (Table 1 in Reles and Friede 1991), the filament density falls into an even narrower range of 125–186 neurofilaments per μm2. Neurofilaments help to regulate the axonal diameter (Sect. 4.1.4).
Found singly or in groups and aligned in parallel to the axon, microtubules measure 25 nm in external diameter and may extend as far as 350 μm. Their cross-sectional density diminishes in larger axons (Friede and Samorajski 1971; Ohnishi et al. 1976): 10–40 tubules per μm2 in myelinated fibers and 50–100 tubules per μm2 in unmyelinated fibers (Berthold 1978; Ohnishi et al. 1976). Microtubules are a fundamental component of the fast axonal transport system, serving as the “track” along which material moves (vide infra). This function probably relates to the close association of tubules with cytoplasmic aggregates and membrane-bound organelles.
Microtubules and neurofilaments form the cytoskeleton of the axon. Although previously thought to be largely fixed in position within the axoplasm (Nixon 1992) and interconnected by a lattice of trabeculae and bridges aligned perpendicular to their long axis (Hirokawa 1982; Schnapp and Reese 1982), recent studies (Brown 2013; Brown and Jung 2013) suggest that neurofilaments are dynamic. Motion in axons along microtubules is bidirectional (but anterograde movement predominates); neurofilaments cycle repeatedly between moving and pausing states throughout their journey along the axon (Brown 2013).
4.1.1.2 Mitochondria
Axonal mitochondria are 0.1–0.3 μm in diameter and 0.5–8 μm in length, with basic ultrastructural features no different than those of mitochondria elsewhere in the body. As with microtubules, the mitochondrial density decreases (in cross sections) with increasing axon diameter. One might expect to see 1–2 mitochondria per cross section in an ordinary unmyelinated fiber and 0.1–0.7 mitochondria per μm2 of axon cross section in myelinated fibers. Density is increased severalfold in the nodal and paranodal segments.
4.1.1.3 Vesicles, Cisternae, and Membranous Tubes
The axon contains various membrane-bound structures: some seemingly empty or multivesicular, some containing granular material, and others having osmiophilic lamellated contents. Most of this membranous material is the axoplasmic reticulum, a meshwork of interconnected tubulovesicular structures contiguous with, but different from, the endoplasmic reticulum of the cell body (Lindsey and Ellisman 1985; Tsukita and Ishikawa 1976). Other vesicles and tubular cisternae, ranging from 50 to 250 nm in diameter, originate from the perikaryal Golgi apparatus and endoplasmic reticulum and are involved in the transport of materials along the axon (Lindsey and Ellisman 1985; Schnapp and Reese 1982; Tsukita and Ishikawa 1976). Most concentrated at the distal paranodal region, dense lamellar bodies and multivesicular bodies probably represent lysosomal or pre-lysosomal organelles in retrograde transport towards the cell body (Lasek and Katz 1987) (Figs. 4.2 and 4.3b).
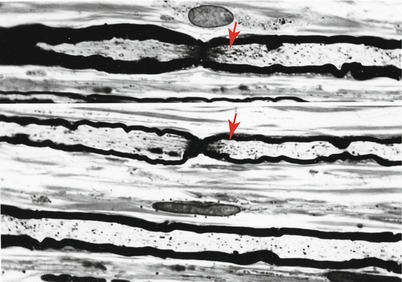
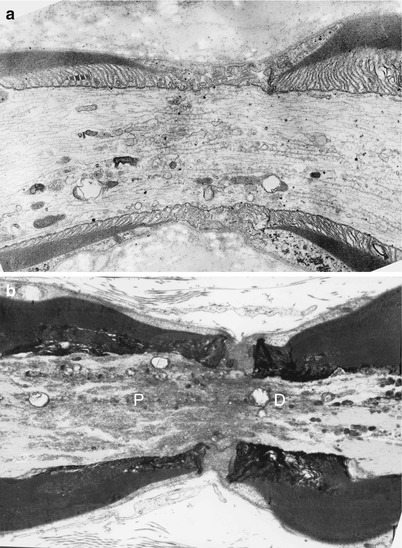
Fig. 4.2
Normal axon. Longitudinal sections of sural nerve show three MFs. Note condensation of axoplasm at nodal and paranodal regions (arrows). (One micron thick plastic section, 1,000×)
Fig. 4.3
Normal axon. Longitudinal section through node and paranodes of thin (a) and thickly (b) myelinated fibers. Note condensation of axoplasm and clustering of organelles. In (b) dense bodies accumulate in the distal paranodal area (D), while tubulovesicular organelles accumulate in the proximal paranodal area (P) (Magnification a 12,840×; b 11,000×)
4.1.1.4 Granular Material
Glycogen appears as approximately 30 nm electron-dense granules. The amount normally found within the axons is scanty and varies between species (Berthold 1978; Zelena 1980). A much finer granular material sometimes accumulates in the nodal segment of the axon and gives it a characteristic darker staining on both light and electron microscopic examination (Fig. 4.3b).
4.1.2 Organization of Axonal Structure
Although the axon is a single, sometimes branched, continuous tube extending from cell body to nerve terminal, myelinated axons do have a periodic architecture as a result of their intimate interaction with segmentally organized Schwann cells. Axon internodal diameter (excluding the myelin sheath) ranges from 1 to 7 μm for myelinated axons and 0.1–3 μm for unmyelinated axons in the human sural nerve (Behse 1990). The composition and organization of axonal contents vary depending on whether nodal or intermodal regions are examined.
The internodal organization of axonal contents is relatively simple: Neurofilaments and microtubules align longitudinally and relatively homogenously and intermingle with tubulovesicular structures and the occasional mitochondrion (Fig. 4.1a, b). A normal internode is 200–1,800 μm in length, roughly proportional to axon diameter.
The node of Ranvier (Fig. 4.1c–e) is the unmyelinated axon segment between two Schwann cells and their myelin sheaths, which is typically 1 μm in length. On either side of the node are the paranodal segments, 10–75 μm in length. Approaching the node, an axon loses its rounded contour and develops grooves that appear imposed by the invagination of the overlying myelin (Fig. 5.5b). These grooves taper towards the nodal segment, where the axon diameter decreases to 50 % or less of its value at the internode (Fig. 4.2). The degree of nodal constriction is proportionally greater for large fibers than for small fibers (Reles and Friede 1991). The constricted axon segment is 5–10 μm in length, with the central 1 μm being the unmyelinated true node of Ranvier (Rydmark and Berthold 1983) and the peripheral 3–5 μm on either side being the site where myelin loops attach to the axon (Figs. 4.1e and 4.3a).
In the nodal–paranodal region microtubules and membrane-bound organelles become concentrated centrally, the former at a density several times greater than that seen in internodal cross sections. Neurofilament density does not change significantly along the axon (Berthold 1978; Reles and Friede 1991). Tubulovesicular profiles assume a longitudinal rather than reticular pattern (Fig. 4.3a, b). Retrogradely transported organelles (dense lamellated bodies and multivesicular bodies) tend to accumulate at the distal half of the nodal and paranodal axon, while anterogradely transported organelles (especially tubulovesicular elements) accumulate at the proximal end (Berthold et al. 1993) (Fig. 4.3b). Both types of organelles occur at far greater concentrations at the node of Ranvier than in the internode. Light microscopy reveals a “veil” of toluidine blue-staining substance in approximately 50 % of nodal–paranodal segments (Figs. 4.1e and 4.2). The ultrastructural correlate of this is a diffuse electron-dense granular material composed of particles 5 nm in diameter (Berthold et al. 1993).
In contrast to the segmental organization of myelinated fibers, axons of unmyelinated fibers have same appearance throughout their length, containing neurofilaments, microtubules, approximately one mitochondrion per cross-sectional view, and relatively infrequent tubulovesicular elements and dense bodies.
4.1.3 Axonal Transport
The bulk of neuronal protein synthesis occurs in the perikaryon rather than the axon. Recent studies have, however, shown that ribosomes, initiation and elongation factors, transfer and messenger RNAs, as well as proteins and microRNAs involved in the regulation of mRNA stability and translation are located in the axon. Locally synthesized axonal proteins include cytoskeletal proteins, protein chaperones, metabolic enzymes, some membrane proteins, and secreted proteins. It is likely that local axonal synthesis has an important and dynamic role in local structure and maintenance, synaptic function, and reactions to injury; however, by itself, local synthesis is insufficient for axonal maintenance. Although mRNA was once thought to be excluded from the axonal compartment, the existence of protein synthesis in growing or regenerating axons in culture is now generally accepted. However, its extent and functional importance remains a subject of intense investigation. Furthermore, evidence of mRNA axonal transport and local gene translation in vivo has been shown in axons of the developing zebra fish (Danio rerio) embryo, with frequent accumulation at the growth cones. Additionally, while supporting cells might provide some of the axon’s metabolic needs (Gainer et al. 1977), this contribution is insufficient by itself, requiring that most axonal material must be transported centrifugally from the cell body over long distances. For several decades investigators have appreciated the existence of a special mechanism for this purpose (Griffin and Watson 1988; Ochs and Brimijoin 1993), recently reviewed in detail (Brown 2013).
The purpose of axonal transport is not always to move a cargo from the cell body directly to a destination at the most distal end of the axon, rather some cargos (e.g., mitochondria) are functionally active during their transit providing long stretches of axons with an ongoing energy supply. Axonal transport functions dynamically, recruiting and redistributing these cargos in response to the changing physiologic and metabolic needs of the axon.
Axonal transport mechanisms have traditionally been organized on the basis of their direction (anterograde and retrograde) and speed (fast or slow) (Brown 2013; Lasek et al. 1984; Guzik and Goldstein 2004). The prototype examples include:
1.
Fast anterograde transport: (50–400 mm/day) carrying Golgi-originating vesicles containing membrane-spanning/membrane-anchoring proteins including neurotransmitters to the nerve terminal and axon.
2.
Fast retrograde transport: (up to 200 mm/day) carrying organelles typical of endocytic, lysosomal, and autophagosomal pathways to the perikaryon. Many are multivesicular bodies containing a variety of cytosomes which may be re-outfitted or destroyed in the perikaryon. Other activated complexes (e.g., Trk/neurotrophin aggregates) are internalized by endocytosis at nerve terminals and sorted into signaling endosomes, which function as carriers to the nucleus where they modulate gene expression. Local synthesis of specific signaling and regulatory proteins in response to axonal injury may initiate a damaged neuron’s injury response.
3.
Slow anterograde transport (possibly as macromolecular complexes) of which there are two main types:
Slow component a (SCa): (0.2–1 mm/day) SCa is composed primarily of neurofilament proteins, tubulin, spectrin, tau protein, and calcium/calmodulin-dependent protein kinase IIb (Brown 2013).
Slow component b (SCb): (about 2–8 mm/day) SCb contains hundreds of non-membranous cytosolic proteins including cytoskeletal proteins (actin, tubulin, cofilin), motor proteins (dynein, dynactin, and myosin), metabolic enzymes (aldolase, creatine phosphokinase, enolase, etc.), and chaperone proteins such as heat shock protein hsp70 (Brown 2013).
Investigators have clarified much of the machinery of fast axonal transport in recent years (Sheetz et al. 1989; Brown 2013). Vesicles can be directly imaged moving along paths demarcated by microtubules at fast anterograde and retrograde rates (Kachar et al. 1987). Deprivation of energy or calcium prevents fast transport, as do agents that depolymerize microtubules. Axoplasmic motor proteins important in axonal transport have been identified: kinesin for fast anterograde and dynein for fast retrograde transport. These proteins have a hinge region and bind to both cargo organelles (tail regions) and microtubules (head region) and, using their ATPase activity, move along microtubules in the presence of ATP (Sheetz et al. 1989; Vallee et al. 1989). Experimental data thus provides strong evidence that fast axonal transport is performed using a microtubule railway upon which a cytoplasmic peptide motor carries vesicles and their contents to the appropriate destination via a calcium- and energy-dependent mechanism. In support of this, mutations in a critical region of β-tubulin (TUBB3) or a motor adaptor protein KIF1Bβ both result in neuropathy (Niwa et al. 2013; Brown 2013). Although the retrograde and anterograde fast transport mechanisms have much in common, specific inhibitors can affect them differently (Edstrom et al. 1988; Guzik and Goldstein 2004).
Slow axonal transport carries cytoskeletal components (neurofilaments predominantly anterogradely) and probably governs the rate of axonal growth and regeneration (Black and Lasek 1979). The classic “structural hypothesis” of slow transport paints a picture of the axonal cytoskeleton composed of tubules and filaments, assembled in the cell body and moving ponderously forward as a whole at a constant rate (Lasek et al. 1984). A competing “unitary hypothesis” suggested that the cytoskeleton is stationary and that its components are transported distally in an unassembled state and assembled locally (Ochs et al. 1989; Bamburg 1988). Recent work (detailed in Brown 2013) suggests that fast and slow transport both take place in association with microtubules; however, they differ in the time the cargo stays in contact with the highway, which has been called the “duty ratio” (the proportion of the time that the cargo spends moving). Neurofilament movement is fast but intermittent, with each filament spending most of its time pausing between short bursts of rapid movement (Tang et al. 2013). Rather than cross-linked neurofilaments and microtubules, the axonal environment may more accurately behave as a polymer solution. Microtubules (all orientated with their minus ends pointing proximally) and microfilaments (relatively flexible, two-stranded, 5–7 nm filamentous polymers of actin capable of multidirectional orientation) serve as the tracks for long-range axial movements in axons and short-range motion, respectively. Neurofilament polymers themselves move along microtubule tracks. However, microfilament myosin motors may influence the long-range transport behavior of mitochondria by delivering these organelles to and from their microtubule tracks. Most motors interact with their cargos via adapter proteins which bind to receptors on the cargo. A single motor may often be called on to interact with multiple different cargoes.
4.1.4 Regulation of Axonal Diameter
Neurofilaments play a central role in determining axonal diameter (Gold et al. 1985; Hoffman et al. 1984, 1987). This conclusion correlates with the observation that localized accumulation of neurofilaments constitutes the substrate of focal axonal swelling and that impairment of neurofilament translocation to the distal axons underlies at least some types of axonal atrophy (vide infra). It is now thought that sidearms produced by NFM and NFH proteins present on neurofilaments preserve space between adjacent filaments and help maintain axonal size, which is critical for conduction velocity (Brown 2013). Additionally, a rigid cross-linked network of neurofilaments would likely retard the movement of other axonally transported cargos. In mutant animals lacking neurofilaments, axons fail to attain their normal caliber and have delayed conduction velocities.
4.2 Axonal Degeneration
4.2.1 Distal Axonopathy
4.2.1.1 Evolution of the Concept
Neuropathy resulting in degeneration of the most distal parts of the axon with progression proximally towards the cell body, i.e., “dying-back” neuropathy, is a common pattern in neuropathies accompanying acrylamide, diabetes, acquired immunodeficiency syndrome (AIDS), alcohol, arsenic, thallium, uremia, isoniazid, and many other medications and organic solvents (Spencer and Schaumburg 1977; Cavanagh 1979; Raff and Whitmore 2002). Superficially, the concept is a simple one: “[If] the neuron were diseased, its trophic function might be impaired, a condition that could result in damage to those regions furthest removed from the source of their trophic input, that is, the distal nerve” (Spencer and Schaumburg 1977). One corollary of this idea is that larger diameter axons, which have a greater volume of axoplasm, and thus presumably higher metabolic demands, would show changes earliest. A second theoretical consequence is that the most distal central nervous system projections of neurons might show similar alterations to those seen in distal peripheral nerve fibers (central–peripheral distal axonopathy).
Clinically, it is noteworthy that the longest peripheral nerve fibers are sensory, i.e., those providing sensation to the most distal aspect of the lower limbs. Moreover, dissociation occurs between the sensory modalities carried by fibers of various sizes: Small myelinated and unmyelinated axons carry sensations of pain and temperature, and large myelinated fibers carry vibration and kinesthetic sensation. Thus, one might expect that in a “dying-back” process the earliest manifestations result from large fiber sensory loss that a “stocking–glove” symmetrical neuropathy would be the typical pattern and that histological studies would demonstrate a relatively more severe loss of large myelinated fibers. Indeed this is the most common clinical and histological disease pattern seen in axonal polyneuropathies.
The excellent correlation between the theory and reality of clinical and pathological observations served to give “dying-back” axonopathy a special place among categories of peripheral nerve disease. In uremic neuropathy a clear proximodistal gradient of nerve disease severity takes place: atrophy appears as the primary manifestation of axonopathy, with degeneration of the nerve fibers occurring in the most distal segment (Dyck et al. 1971). Similar changes have been demonstrated in other neuropathies.
In some models of toxin-induced axonal degeneration, however, careful ultrastructural and teased fiber studies revealed that the pathological changes do not always occur in a “dying-back,” axon length- and diameter-dependent pattern and that axonal atrophy does not necessarily take place. Instead, axonal swelling and myelin retraction occurred in multifocal sites, more prominent in but not necessarily confined to the most distal and largest axons (Spencer and Schaumburg 1977; DeRojas and Goldstein 1990). In some studies axonal swellings tended to occur in the proximal paranodal segment of a given internode. Teased fiber studies suggest that focal axonal swelling might cause demyelination and then resolve, allowing remyelination and thus permitting secondary demyelination/remyelination (Griffin and Price 1981).
Originally suggested by Spencer and Schaumburg (1977) as a replacement for the term “dying-back” neuropathy, “distal axonopathy” encompasses both the typical (uremia-like) and less typical (hexacarbon intoxication-like) varieties of length-dependent axonal degenerative processes. The term can probably apply to nearly all axonal metabolic polyneuropathies, whether due to toxins, endocrine disturbances, organ failure, inherited defects, or nutritional deficiency. Ischemic neuropathies and neuropathies resulting from local damage to nerves (inflammation, amyloid) are not considered distal axonopathies. However, even in these settings similar features occur (Said et al. 1984). The axonopathic ultrastructural changes discussed below are a reflection of the fact that the axon’s metabolic machinery is disturbed and represent nonspecific indicators of a “sick” axon or neuron.
4.2.1.2 Pathogenetic Mechanisms
The pathophysiological mechanisms underlying distal axonopathy have been a subject of intense research for a number of years (Spencer and Schaumburg 1977). The classic notion has been that metabolic function in the cell body, responsible for production of substances essential for survival of the axon, is impaired in some unspecified fashion. Even in normal nerves many transported substances show a proximodistal decreasing gradient in their concentration, presumably on the basis of ongoing degradation and/or utilization of transported materials as they proceed distally (Miller and Spencer 1985). Thus, a defect in production of vital macromolecules would show up first in the distal axon. Attempts to study cell body metabolic activity by assessment of amino acid uptake yielded equivocal results (Spencer and Schaumburg 1977). One hypothesis, derived from the acrylamide toxic neuropathy model, is that interference with glycolysis caused depletion of high-energy groups needed for axonal maintenance (Spencer et al. 1979). Subsequent investigation, however, has called into question the significance of these changes in axonal metabolism (Miller and Spencer 1985).
One alternative to the concept of the cell body as the site of initial disturbance in distal axonopathy is the possibility that it results from a local axonal defect. For example, a circulating toxin might cause a deficiency of, or demand for, a critical macromolecule throughout the length of the axon. Since such a macromolecule would likely arrive from the perikaryon and may occur in lesser concentrations more distally in the fiber (Miller and Spencer 1985), distal parts of the axon would, in this paradigm, suffer the greatest effects (Spencer and Schaumburg 1977).
Both of the above proposed mechanisms for distal axonopathy depend on the notion of transport of essential substances from the cell body to the distal axon. In recent years, attention has increasingly focused on defects of axonal transport as a cause of distal axonopathy. Axonal swellings, a characteristic feature of some human neuropathies including hexacarbon toxicity and giant axonal neuropathy, result from focal accumulations of neurofilaments. This accumulation suggests that impairment of filament transport might underlie the neuropathy. Similarly, distal axonal atrophy, a ubiquitous component of numerous inherited, toxic, and metabolic neuropathies, might be related to impaired production or centrifugal transport of neurofilaments (Hoffman et al. 1984, 1987). Data have at times been conflicting (Spencer and Schaumburg 1977), but potential specific examples of how axonal transport impairment leads to neuropathy include alterations in slow transport (iminodipropionitrile, Griffin et al. 1978) or fast transport (hexacarbons, Mendell et al. 1977) or a disturbance of the microtubules that form the backbone of the transport system (vincristine, colchicine, podophyllotoxin) (Paulson and McClure 1975; Sahenk et al. 1987). Failure of retrograde transport or of the normal mechanism by which a proportion of the membrane and protein delivered distally returns proximally (“turnaround defect”) might result in distal accumulation of abnormal, possibly toxic, materials. This situation has been demonstrated in an acrylamide neuropathy model where focal accumulation of particulate organelles constitutes a prominent early feature (Miller and Spencer 1985; Chretien et al. 1981).
Recent studies have proposed additional mechanisms for distal axonopathy, in this case in the setting of diabetic neuropathy. Dying-back axonal degeneration has been proposed to reflect the activation of a self-destruct program in the distal parts of an axon in response to a neuronal insult (Raff and Whitmore 2002) providing a neuron with several choices. One choice is to prevent neuronal death using a controlled self-protective reaction to a metabolic burden, conserving resources so that the preterminal axon and axon terminals can be regenerated at a later time when the neuropathic stress has abated (Raff and Whitmore 2002). Mitochondrial pathology, perhaps reflecting injury secondary to local oxidative stress to the mitochondrial genome or difficulty with mitochondrial calcium homeostasis, may underlie distal axonopathy. In diabetic neuropathy Fernyhough and colleagues (Chowdhury et al. 2013) proposed that the distal axon has a higher and more fluctuating demand for ATP resulting from collateral sprouting and synaptic plasticity (possibly involving cycles of degeneration/regeneration), not paralleled in the perikaryon. Abnormal mitochondria with diminished spare respiratory capacity in diabetic animals are thought to be less capable of adapting to high peaks of ATP demand within the distal axon and result in its degeneration.
Although experimental observations are often based on the study of neurotoxins, similar alterations in axonal transport contribute to a wide variety of neuropathies. Even the “neuropathy of aging” might result from a gradual age-related reduction in transport effectiveness (McQuarrie et al. 1989).
4.2.2 Pathological Alterations in Axonopathy
Studies in animal models and human pathology have provided important insights into the spatiotemporal sequence of distal axonopathy (Spencer and Schaumburg 1977; Prineas 1969a, b; Sahenk and Mendell 1980). Most changes of distal axonal degeneration are nonspecific, but some etiologies have specific patterns. For example, acrylamide and the hexacarbon neuropathies tend to produce neurofilamentous accumulations and axonal swelling; TOCP and zinc pyridinethione produce tubulovesicular aggregates; and thallium intoxication results in mitochondrial abnormalities. However, we cannot catalog all the various combinations of ultrastructural changes that investigators have observed in the numerous animal toxic models and human ultrastructural examinations conducted to date. Such an undertaking would not prove diagnostically useful in the practice of peripheral nerve pathology. On the contrary, the discussion below will emphasize the nonspecificity of ultrastructural changes seen in axonal degeneration. Similar changes occur in axonal degeneration in the CNS (Lampert 1967). No single change is diagnostic, or even necessarily indicative of pathology, as the wide variation of normal appearance, age-related changes, and artifacts induced by the trauma of nerve biopsy and fixation can be misleading. Many of the same alterations are seen in Wallerian degeneration following mechanical or ischemic nerve transection.
4.2.2.1 Axonal Swelling and Filamentous Accumulation
Neurofilament accumulation is the ultrastructural correlate of axonal swellings described to varying degrees in a wide range of human and experimental neuropathies, including giant axonal neuropathy, hexacarbon intoxications, disulfiram, acrylamide (Davenport et al. 1976; Prineas 1969b), iminodipropionitrile (Griffin et al. 1978), carbon disulfide (Gottfried et al. 1985), and one CMT-2 pedigree (Vogel et al. 1985). Investigators also have observed filament accumulations in a case of B12 deficiency (Schochet and Chesson 1977), in amyloidosis (Hanyu et al. 1989; Jedrzejowska 1977), and in misonidazole toxicity (Urtasun et al. 1978). Typically, axons demonstrate a fusiform swelling to several times their normal diameter, and electron microscopy reveals accumulations of neurofilaments arrayed in swirling masses in various planes of orientation. Other organelles are diminished in numbers or displaced. The focal swellings often involve only part of an internode. In hexacarbon neuropathies, these swellings often occur just proximal to a node of Ranvier, their frequency increasing in a proximodistal gradient along the nerve fiber (Spencer and Schaumburg 1977). Myelin overlying the axonal swellings may become thinned, and paranodal myelin retraction and focal demyelination may occur, which is seen best with teased fiber analysis. This axonal change may at some stage still be reversible. As the axon dilates and narrows, it may shed its myelin, resulting in secondary segmental or subsegmental demyelination and remyelination and, if the process is chronic, onion-bulb formation (Spencer and Schaumburg 1977; Griffin and Price 1981).
A rare swollen axon with filamentous accumulations is an infrequent but nonspecific finding that we have seen in cases with diagnoses as varied as CIDP, CMT-2, or granulomatous neuropathy (Fig. 4.4a, b). However, when such axons are found in greater numbers, typically several per fascicle, this finding has diagnostic implications (Table 7.12). Increased neurofilament density without prominent axonal enlargement can occur in vincristine and cisplatin toxicity (Gastaut and Pellisier 1984; Wulfhekel and Dullmann 1972). We have examined a biopsy in porphyric neuropathy where focal swellings of unmyelinated axons with filament accumulations appeared frequently (Thorner et al. 1981).
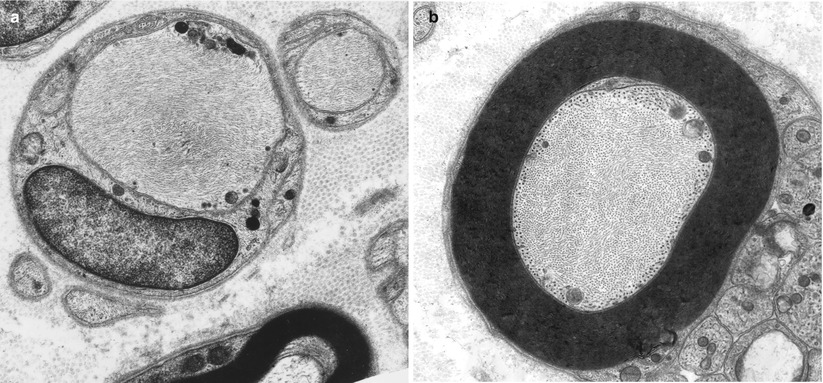
Fig. 4.4
Axonopathy. Intra-axonal filamentous aggregates are nonspecific findings in some chronic neuropathies. Note segregation and marginalization of microtubules in b (Magnification, a 15,390×; b 20,000×)
The pathological significance of axonal swellings in intramuscular nerves is uncertain (Barron and Heffner 1978; Wolfhart 1957) given that swelling occurs in a high proportion of normal human intramuscular nerves and in neuropathies presently thought to be not of the distal axonopathy variety (Alderson 1992).
4.2.2.2 Axonal Atrophy
Axonal atrophy is more difficult to recognize than axonal swelling because the changes may be subtle, require quantitative techniques for adequate analysis, and can resemble an artifact. An axon which is clearly inappropriately small for its myelin sheath, often lying in the center of an apparently empty space surrounded by myelin (Fig. 4.5a, b), indicates evidence of atrophy. It is hard to see how the myelin sheath, ordinarily in such intimate anatomical and physiologic contact with the axon, could maintain its structural integrity when separated from its axon in the fashion such micrographs suggest. In some instances, organelles are identified suggesting that the “empty” space actually represents a swollen adaxonal Schwann cell cytoplasmic process (Fig. 4.5a). Convincing demonstration of atrophy requires an intensive quantitative study. Dyck and colleagues have provided such studies for a variety of neuropathies including those of uremia, Friedreich’s ataxia, some paraproteinemic neuropathies, and permanent axotomy (Dyck et al. 1971, 1981; Dyck and Lais 1973; Ohi et al. 1985). Suggestive data also exists for some toxic neuropathies in humans and animals including ethylene oxide (Schroder et al. 1985), cisplatin (Gastaut and Pellisier 1984), hereditary motor and sensory neuropathy (CMT) types 1 and 2 (Dyck et al. 1993b; Yasuda et al. 1990), hereditary sensory and autonomic neuropathies (HSAN) (Dyck 1993), and HIV-associated neuropathy (Fuller et al. 1990).
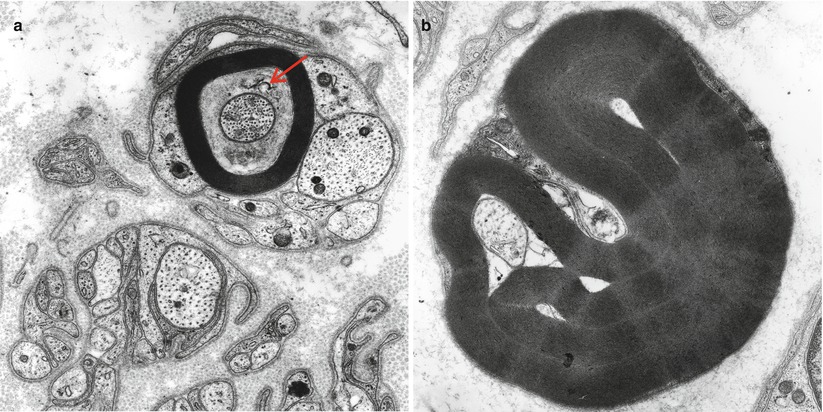
Fig. 4.5
Axonopathy. Axonal “atrophy” seen in a case of nonspecific chronic neuropathy (a) and a case of HNPP (b). Note organelles in adaxonal space (arrow, a) (Magnification, a 20,000×; b 12,000×)
Conclusive identification of axonal atrophy requires demonstration of a shift of fiber diameter–frequency histograms to the left and a decrease in the slope of axonal area/diameter vs. myelin thickness plots, showing that the axon is inappropriately small for its myelin thickness (Ohi et al. 1985). Myelin remodeling often follows axonal atrophy and manifests as excessively wrinkled myelin with numerous infolded loops, bubbles, or fissures (Dyck et al. 1984). Secondary demyelination, as demonstrated by teased fiber studies, often follows.
Neurofilament numbers largely regulate axonal diameter, and neurofilament density is relatively constant within different nerves (vide supra). Thus, a demonstration that this density is not significantly altered would further support the claim that the observed axonal shrinking is not an artifact (Friede 1971; Yasuda et al. 1990). On the other hand an “atrophic” myelinated axon with a markedly increased neurofilament density should be regarded with skepticism. Other organelles, including mitochondria, tubulovesicular profiles, and microtubules, increase in density when an axon undergoes atrophy (Friede 1971).
4.2.2.3 Mitochondrial Abnormalities
Mitochondria may show a variety of changes including focal accumulations (Fig. 4.6a), enlargement, excessive or disorganized cristae, increased electron density of the matrix, accumulation of coarse granular (probably glycogen) or fine granular osmiophilic material, varying degrees of loss of structural integrity, and paracrystalline or amorphous electron-dense inclusions. These changes have been described in thallium intoxication (Spencer and Schaumburg 1977), uremia (Dyck et al. 1971), alcohol abuse (Tredici and Minazzi 1975), vitamin E deficiency (Schochet 1971), Tangier disease (Dyck et al. 1978), ataxia-telangiectasia (Gardner and Goodman 1969), and numerous toxic agents. These toxic substances include acrylamide, hexacarbons, INH, cisplatin, and vincristine (Bradley et al. 1970; Schlaepfer and Hager 1964; Spencer and Schaumburg 1977; Prineas 1969b; Gastaut and Pellisier 1984). Mitochondrial alterations are also seen in mitochondrial cytopathies (Chap. 21) but more prominently in Schwann cells than in axons. Specific changes such as crystalline inclusions are vanishingly rare.
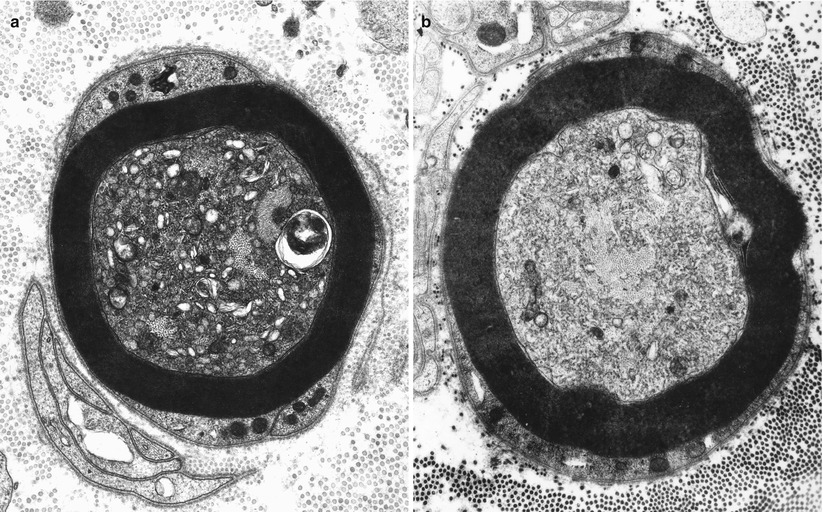
Fig. 4.6
Axonopathy. Alteration of axoplasm with segregation of neurofilaments and pleomorphic vesicular accumulations. In (b) most of the axoplasm displays tubulovesicular profiles with a bundle of neurofilaments at the center. Note similarity to neuroaxonal dystrophy (Figs. 19.26 and 19.27). (Magnification, a 21,546×; b 13,760×)
4.2.2.4 Aggregation of Membranous Organelles and Dense Bodies
In experimental organophosphate intoxication in the cat, Prineas (1969a) observed accumulation of abnormal membrane-bound spaces, arranged as vesicles, tubules, and flattened sacs. These spaces also occur in dimethylaminopropionitrile toxicity (Pestronk et al. 1980), and they are a nonspecific feature of axonal neuropathies, including familial hereditary motor and sensory neuropathy (Yasuda et al. 1990), thiamine deficiency (Takahashi and Nakamura 1976), isoniazid toxicity (Ochoa 1970), amyloidosis (Jedrzejowska 1977), and Tangier disease (Dyck et al. 1978) (Fig. 4.6a, b). Similar considerations apply to accumulation of “dense bodies” (Prineas 1969b; Dyck et al. 1978; Meier and Bischoff 1977; Tredici and Minazzi 1975). Organelles may appear segregated, with domains containing masses of neurofilaments adjacent to regions containing only membranous organelles (Fig. 4.6a, b). With focal nerve ischemia, axonal swelling and accumulation of organelles frequently appear in regions proximal, and to a lesser extent distal, to the site of axonal disruption (Korthals et al. 1978). In neuroaxonal dystrophy, the essential pathological alteration is the accumulation of tubulovesicular profiles with focal congregations of mitochondria, filaments, glycogen and various dense bodies, granules, and vacuoles (Figs. 19.26 and 19.27).
4.2.2.5 Schwann Cell–Axon Networks
Schwann cells participate in the clearance of damaged organelles and removal of axonal debris (Spencer and Thomas 1974). A ridge of Schwann cell cytoplasm evaginates into the axon near a collection of axonal organelles which often show atypical features (enlarged mitochondria, clear vesicles, multivesicular or dense membranous bodies, or collections of neurofilaments). This ridge progressively elongates and thins out, tending to surround the abnormal organelles. It then folds and fuses within itself, sequestering aggregates of abnormal axoplasmic contents. The outer sheath of this Schwann cell evagination is maintained, but inner subdivisions fade away, so that the sequestered axoplasmic debris enters the Schwann cell. In cross-sectional anatomical studies, this process results in a honeycombing appearance at the periphery of the axon, with aggregates of granular debris trapped within the individual honeycomb cells (Fig. 4.7a, b). These networks usually appear in paranodal regions, most frequently in large myelinated fibers and only rarely in unmyelinated fibers (Spencer and Thomas 1974).
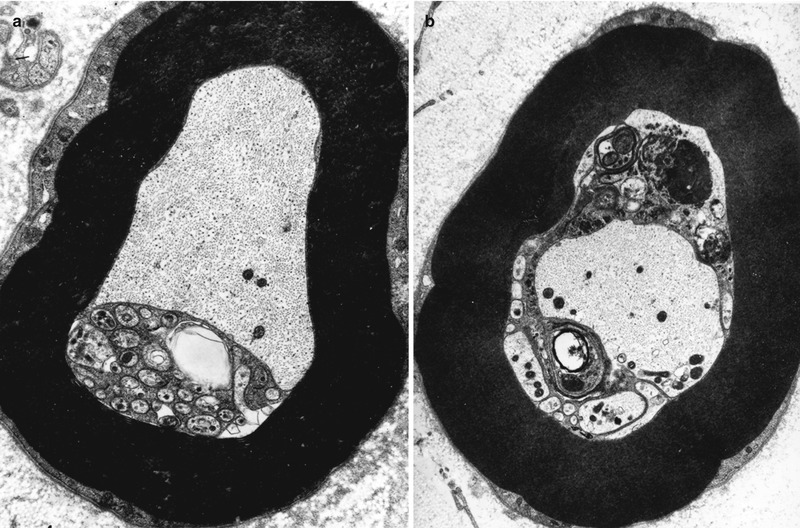
Fig. 4.7
Axonopathy. Schwann cell–axon networks are shown in two MFs. Pockets of axoplasm containing degenerate organelles are incorporated into an inner evagination of Schwann cell cytoplasm (Magnification, a 10,944×; b 7,820×)
Normal axons may demonstrate such Schwann cell–axon networks, almost exclusively in small numbers in the paranodal area and not in association with abnormal myelin configurations. Hexacarbon, acrylamide, and thallium intoxications, this observation occurs in axonopathy of various etiologies, including CMT-2 (Yasuda et al. 1990), isoniazid intoxication, and thiamine deficiency in rats (Collins et al. 1964; Schlaepfer and Hager 1964); experimental hexacarbon toxicity, acrylamide and thallium intoxication (Spencer and Thomas 1974); cisplatin (Gastaut and Pellisier 1984), lead (Schlenska and Spalke 1975), and sodium cyanate (Ohnishi et al. 1975) toxic neuropathies; and porphyria-, uremia-, or lymphoma-associated neuropathy (Thorner et al. 1981; Ahonen 1981; Vital and Vallat 1987) in humans. Schwann cell–axon networks may also occur proximal to a focal nerve lesion (Spencer and Thomas 1974) and in the neuropathy associated with chronic vascular insufficiency (Farinon et al. 1984). This finding likely represents a nonspecific mechanism by which the Schwann cell clears debris and helps maintain the integrity of the axon under normal and pathological situations (Gatzinsky and Berthold 1990). Overall, this ultrastructural feature is an uncommon one, which has theoretical importance but little role in the differential diagnosis of human peripheral nerve pathology.
4.2.2.6 Other Nonspecific Changes
Axons may show a reduction in the various organelles normally present or an accumulation of membrane-bound granular debris. Workers have observed a host of filamentous, granular, and lamellar inclusions in axons, none of which is specific to any particular disease or even necessarily a sign of pathology (Figs. 4.8a–d, 4.9a, b, and 4.10). Excessive axonal glycogen has been emphasized in hypothyroidism and in diabetes but is a nonspecific finding (Fig. 4.11a, b).
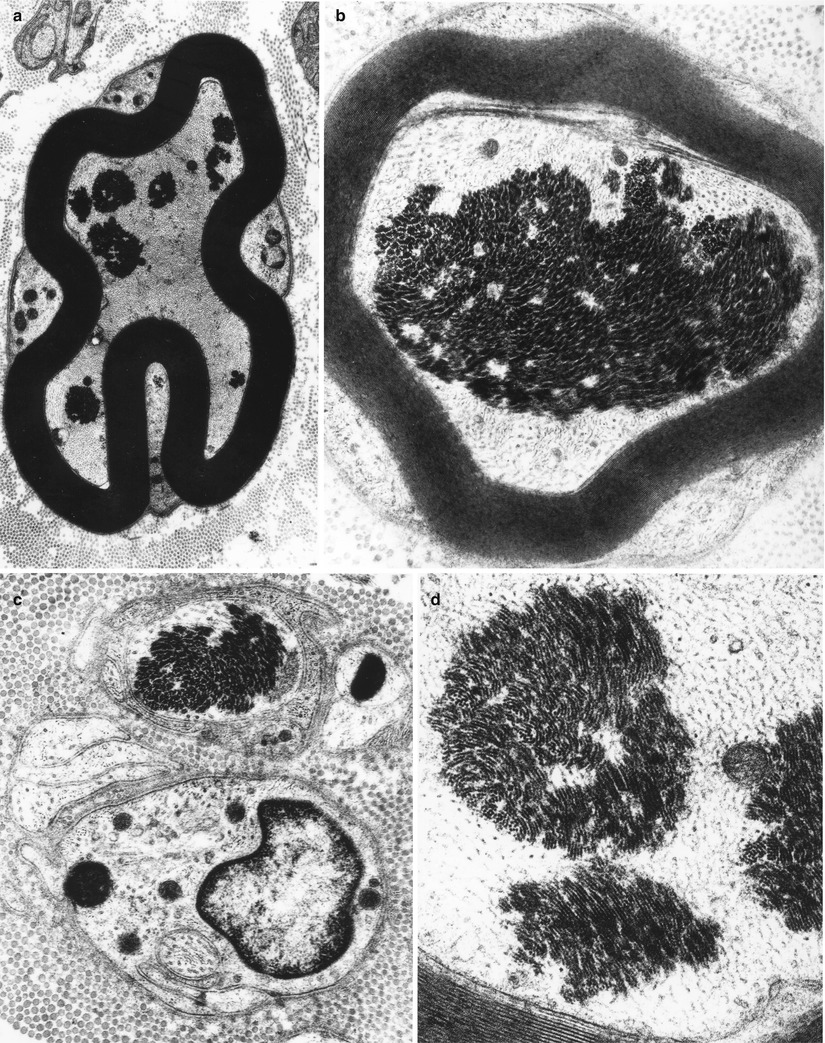
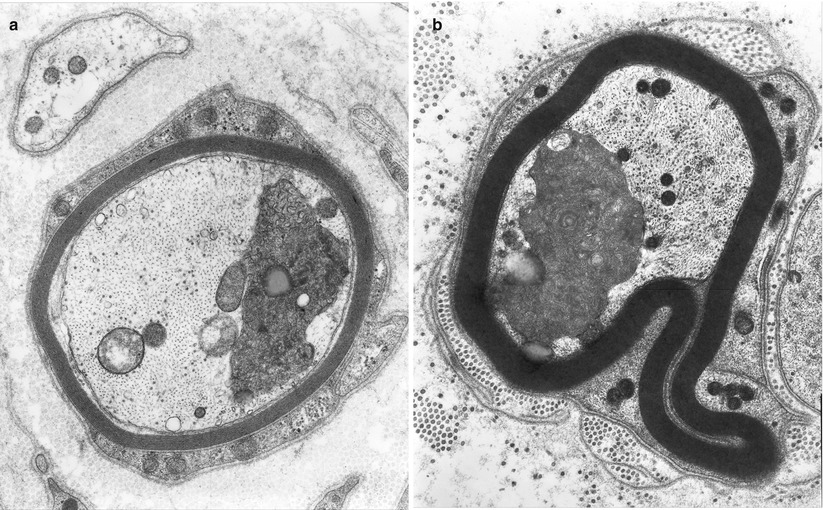
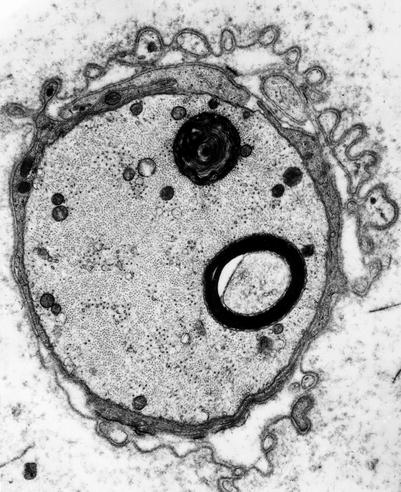

Fig. 4.8
Axonopathy. Intra-axonal aggregates of filaments appear as paracrystalline structures (Magnification, a 8,360×; b 22,344×; c 23,800×; d 42,600×)
Fig. 4.9
Axonopathy. Intra-axonal compact membranolamellar structures are nonspecific (Magnification: a 25,048×; b 26,600×)
Fig. 4.10
Axonopathy. Unusually large intra-axonal myeloid bodies are seen in a chronic axonal neuropathy (Magnification, 15,000×)
Fig. 4.11
Axonopathy. Nonspecific findings in a nerve biopsy include intra-axonal aggregates of glycogen, either membrane bound (arrow, a) or diffusely distributed (*, b) (Magnification, a 9,712×; b 10,260×)
4.3 Wallerian Degeneration
When an axon is transected, most often by trauma or ischemia, a well-studied sequence of events follows as the nerve segment distal to the site of transection degenerates. This process has been named Wallerian degeneration after the pathologist who offered early morphologic observations first emphasized the importance of distal degeneration (Waller 1850). Physiologic studies in man indicate that distal to a transection the axon does not degenerate immediately. Rather, it remains electrically excitable and capable of impulse conduction for as long as 4 or more days, depending on nerve length, with longer distal stumps surviving a few more days (Chaudry et al. 1992). In small experimental animals, the duration of distal stump survival is less, typically 1–2 days. Recent development of animal models of Wallerian degeneration has resulted in rapidly increasing body of literature revealing the complexity of the process (vide infra).
Although the terms “axonal degeneration” and “Wallerian degeneration” are often used interchangeably and may have shared pathogenetic mechanisms, Wallerian degeneration specifically constitutes the microscopic reactions of a nerve segment distal to a site of crush or transection injury. In Wallerian degeneration, histological changes are stereotyped and synchronized compared to neuropathies in which axonal degeneration and regeneration often occur simultaneously.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


