The Floppy Infant
Darryl C. De Vivo
Jahannaz Dastgir
INTRODUCTION
Tone opposes gravity and maintains postural attitude. The term hypotonia refers to a state of low muscle tone. Infants who are hypotonic are often referred to as floppy because their limbs and head hang limply rather than firmly extended when held by an examiner in the prone position. These infants also may show a diminished resistance to passive limb movement when examined. The floppy infant seems to be weak, but the examiner may find it difficult to distinguish between true weakness and apparent weakness due to hypotonia and/or hyperlaxity.
Causes for hypotonia in infants are many, and it is the responsibility of the clinician to create a relevant differential diagnosis for the clinical presentation (Fig. 136.1). The clinician should first focus on the presenting history, noting weeks of gestation, intrauterine drug exposure, perinatal ultrasound abnormalities (e.g., cerebral dysgenesis, reduced fetal movements, oligohydramnios, or polyhydramnios), intrauterine infections (herpes simplex, cytomegalovirus, toxoplasmosis, rubella), and perinatal drug exposure or trauma (global or focal). A notation also should be made on whether the infant had low tone, limb contractures, difficulty breathing, or a weak cry at birth. This information helps determine whether the condition began before, at, or after birth. Hypotonia, emerging days after birth suggests an inborn error of metabolism. Additional history, when evaluating an infant in the first month of life, should include the strength of the infant’s suck and whether the infant chokes with feeds. Potential exposures to Clostridium botulinum (via honey or corn syrup consumption or dirt) are important. Family history may reveal an inherited risk for hypercoagulability (including prior maternal spontaneous abortions), immune-mediated conditions, genetic syndromes (premature death may suggest a metabolic syndrome), neuropathies, myasthenia gravis, or myopathies/muscular dystrophies (developmental delay and weakness).
The aforementioned history will complement a careful neurologic exam that identifies (1) unique dysmorphic features; (2) a highly arched palate or tenting of the mouth (suggestive of congenital tongue weakness); (3) respiratory distress; (4) heart, abdominal, genitourinary, skin (birthmarks, doughy quality), or musculoskeletal abnormalities (congenital scoliosis, kyphosis, contractures, hip dysplasia, torticollis, hyperextensibility of joints); (5) level of alertness; (6) quality of cry; (7) cranial nerve dysfunction (facial weakness, ptosis, limited extraocular movements, tongue fasciculations, tongue atrophy, strength of suck); (8) focal or diffuse weakness; (9) evidence of hypertonicity (increased jitteriness and rigidity of the limbs) or hypotonicity (frog-legged posture while supine, lack of spontaneous movement, scarf sign, tendency to keep the trunk flexed while held in horizontal suspension, slip through); and (10) tendon reflex responses. The pertinent findings on examination, in conjunction with a thorough history, will guide the clinician to whether the infant’s presentation is related to a nonneurologic (structural, toxic, or genetic syndrome) or neurologic etiology that may be central (brain and spinal cord) or peripheral (peripheral nerves, neuromuscular junction, or muscle).
If the clinician believes that the hypotonia is neurologic and central in nature, additional workup would include brain imaging (magnetic resonance imaging [MRI] or computed tomography [CT] scan), electroencephalography (EEG), and cerebrospinal fluid (CSF) studies. If the hypotonia is neurologic and peripheral, confirmatory studies may include electromyography (EMG), nerve conduction studies (NCS), serum creatine kinase (CK) levels, muscle ultrasound or MRI, and muscle and/or nerve biopsy. If the findings from these tests raise concerns for a dystrophy or myopathy, additional processing of the muscle for electron microscopy (to identify distinctive ultrastructural abnormalities), immune staining, or biochemical assays for specific enzyme deficiencies should be performed.
If the clinician finds that the hypotonia is nonneurologic, confirmatory diagnostic workup should address a particular genetic syndrome (chromosomal microarray, whole exome sequencing, or targeted gene sequencing), inborn error of metabolism (blood studies—e.g., glucose, electrolytes, blood gas, ammonium, lactate and pyruvate, carnitine profile, liver function testing, very longchain fatty acids, plasma amino acids, 7-dehydrocholesterol, isoimmune electrophoresis for transferrin; urine studies—e.g., urinalysis, urine amino acids, urine organic acids; CSF studies— e.g., amino acids, lactate, pyruvate, neurotransmitters, pterins, and 5-methyltetrahydrofolate), or structural abnormality (bone films, spinal MRI). These studies complement the newborn screening.
CAUSES OF CENTRAL HYPOTONIA
Central hypotonia refers to decreased tone due to lesions above the anterior horn cells of the spinal cord. Such hypotonia is often symmetric and affects the upper and lower limbs equally. It may also present with additional features of developmental delay (speech, motor, or global), seizures, and/or brisk reflexes.
HYPOXIC-ISCHEMIC ENCEPHALOPATHY
Hypoxic-ischemic encephalopathy (HIE) develops in the setting of perinatal hypoxia. The initial presentation of HIE in the newborn period may be hypotonia in addition to seizures, decreased level of alertness, and/or irritability. Over time, the initial presentation of hypotonia may evolve into increased tone and spasticity.
NEURONAL MIGRATION DISORDERS
Neuronal migration disorders include holoprosencephaly, anencephaly, lissencephaly, pachygyria, polymicrogyria, and/or heterotopias. It should be noted that neuronal migration disorders might be part of a larger syndrome, and some of these syndromes also present with peripheral hypotonia either in the form of myopathy, muscular dystrophy, or neuropathy (e.g., merosindeficient congenital muscular dystrophy, Fukuyama congenital muscular dystrophy, Walker-Warburg syndrome, or muscleeye-brain disease).
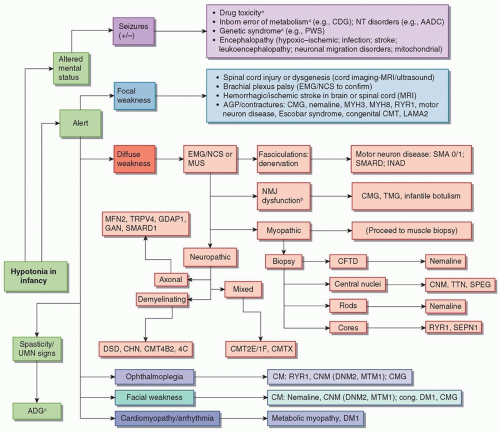 FIGURE 136.1 Differential diagnosis for hypotonia in infants. aMay be associated with pathognomonic dysmorphic features. βMuscle ultrasound (MUS) is not diagnostic for neuromuscular junction dysfunction but can visualize fasciculations and fibrofatty replacement in muscle either due to myopathy, neuropathy, or motor neuron disease. cMay also present with seizures. AADC, aromatic l-amino acid decarboxylase deficiency; ADG, α-dystroglycanopathy; AGP, arthrogryposis; CDG, congenital disorder of glycosylation; CFTD, congenital fiber-type disproportion; CHN, congenital hypomyelinating neuropathy; CM, congenital myopathy; CMG, congenital myasthenia gravis; CMT, Charcot-Marie-Tooth; CNM, centronuclear myopathy; DM1, myotonic dystrophy; DNM2, dynamin 2 gene; DSD, Dejerine-Sottas disease; EMG, electromyography; GAN, giant axonal neuropathy; GDAP1, ganglioside induced differentiation associated protein 1; INAD, infantile neuroaxonal dystrophy; LAMA2, laminin α-2 gene; MFN2, mitofusin 2 gene; MRI, magnetic resonance imaging; MTM1, myotubularin 1 gene; MUS, muscle ultrasound; MYH, myosin heavy chain; MYH3, myosin heavy chain 3 gene; MYH8, myosin heavy chain 8 gene; NCS, nerve conduction studies; NMJ, neuromuscular junction; NT, neurotransmitter; PWS, Prader-Willi syndrome; RYR, ryanodine receptor 1 gene; SEPN1, selenoprotein 1 gene; SMA, spinal muscular atrophy; SMARD1, spinal muscular atrophy and respiratory distress type 1; SPEG, striated muscle preferentially expressed protein kinase; TMG, transient myasthenia gravis; TRPV4, transient receptor potential cation channel, subfamily V, member 4; TTN, titin gene; UMN, upper motor neuron. |
LEUKODYSTROPHIES
Leukodystrophies may also present with a concomitant demyelinating neuropathy (see “Peripheral Neuropathy” section).
Leukodystrophies are disorders of central nervous system (CNS) myelin development and maintenance (divided into disorders of dysmyelination, hypomyelination, or demyelination). They are genetically determined and may present in infancy with hypotonia, encephalopathy, and/or developmental delays.
VASCULOPATHIES: ISCHEMIC OR HEMORRHAGIC
Neonatal stroke may initially present as hypotonia and later evolve into hypertonia with spasticity.
CONGENITAL INFECTIONS
Congenital infections include toxoplasmosis, Treponema pallidum, rubella, cytomegalovirus, herpesvirus, hepatitis B and C viruses, HIV, varicella, and parvovirus B19. These may present with hypotonia in addition to other features of intracranial calcifications or cysts, abnormal liver function, skin rash, hearing loss, encephalopathy, seizures, and/or retinitis or optic nerve atrophy.
INBORN ERRORS OF METABOLISM
Refer to Chapters 134 and 139. Some inborn errors of metabolism may also cause peripheral neuropathy.
GENETIC SYNDROME
Genetic syndromes that most commonly present with hypotonia include Down syndrome, Prader-Willi syndrome, fragile X syndrome, trisomy 18, 1p36 deletion, 22q13 deletion syndrome, 22q11.2 deletion syndrome, DiGeorge/velocardiofacial syndrome, Williams syndrome, trisomy 13, Smith-Magenis syndrome, Sotos syndrome, Lowe syndrome, Smith-Lemli-Opitz syndrome, Wolf-Hirschhorn syndrome, Kabuki syndrome, Cri-du-chat syndrome.
TOXICITY
Intrauterine exposure to drugs such as lamotrigine, lithium, and benzodiazepines have been associated with hypotonia. Antepartum or peripartum maternal exposure to high levels of magnesium sulfate for the treatment of eclampsia and prevention of cerebral palsy has also been associated with congenital hypotonia.
NERVE/CORD INJURY OR STRUCTURAL ABNORMALITY
For discussion of spinal dysraphism/spina bifida, refer to Chapter 133.
Spinal cord injury may be secondary to intrauterine malpositioning or trauma/severe asphyxia during birth (via hypoperfusion between the anterior spinal artery and paired spinal arteries) that subsequently damages the lower motor neurons of the spinal cord. Autopsy studies of fatally asphyxiated neonates demonstrate prominent, ischemic necrosis of anterior spinal cord gray matter. Infants with spinal cord injury often present with hypotonia, weakness, and absent tendon reflexes only in the regions distal to the site of injury. Ultrasound/MRI of the spine also may be used to identify cord ischemia or spinal dysraphism.
CAUSES OF PERIPHERAL HYPOTONIA
Peripheral hypotonia refers to disorders of the motor unit/anterior horn cell, peripheral nerve, neuromuscular junction, and/or muscle. Identifying whether or not there is focality to the weakness often helps with diagnosing its etiology.
FOCAL WEAKNESS: BRACHIAL PLEXUS INJURY
Epidemiology
Brachial plexus injury is variable and ranges from 0.38 to 3 per 1,000 live births in industrialized countries. Birth incidence depends on the type of obstetric care and the average birth weight (strongest predictor) of infants in different geographic regions.
Pathobiology
Shoulder dystocia is a strong predictor for brachial plexus palsy. Vaginal deliveries associated with vacuum extraction or direct compression of the fetal neck during delivery by forceps can cause stretching of the cervical nerve roots (via misdirected traction or hyperextension of the arms) and eventually, brachial plexus injury. A prolonged second stage of labor may also increase the risk.
Clinical Features
Brachial plexus palsies are divided into four categories based on location of injury: (1) upper (Erb palsy, C5-C7, most common palsy presenting with adducted arm that is internally rotated at the shoulder; wrist is flexed and fingers are extended in a “waiter’s tip” posture), (2) intermediate (C7 and sometimes C8 and T1), (3) lower (Klumpke paralysis—C8, T1—poor hand grasp with more intact proximal muscles), and (4) total plexus palsy (C5-C8 and sometimes T1—second most common and the most devastating—claw hand and flaccid arm with no sensation). Additional finding that may be associated with a poor prognosis is Horner syndrome.
Diagnosis
X-rays of the chest, spine, and upper limbs are most important for brachial plexus palsies because they reveal associated injuries (fracture of ribs, transverse process, clavicle, humerus, or phrenic nerve injury). MRI of the upper limb/shoulder and spinal MRI also should be performed to rule out nerve root evulsion. CT myelography may be used if MRI is not possible. EMG/NCS may identify the severity of the neural lesion and detect signs of reinnervation. Serial EMG/NCSs within the first 48 hours after birth will distinguish between prenatal and obstetric causes of brachial plexus palsy. Sensory-evoked potentials can be used to assess the integrity of sensory conduction.
Treatment
For brachial plexus palsy, supportive therapy to maintain passive range of motion and muscle strength is recommended. The optimal timing of surgical exploration remains controversial but most recommend intervention in the first 6 months of life if there is no evidence of clinical recovery.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


