and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
9.1 Guillain–Barré Syndrome
Guillain–Barré syndrome (GBS, reviewed in Arnason and Soliven 1993; Ropper et al. 1991; McFarlin 1990; and, most recently, in Rinaldi 2013), the most common cause of acute–subacute neuropathy, has an incidence of 1.5–2 cases per 100,000 throughout the world (Arnason and Soliven 1993). GBS is characterized by the development of a rapidly progressive paralytic syndrome, and although often thought of as a motor disease, sensory and autonomic symptoms may occur before motor dysfunction. Despite the presence of several variants, most cases are so typical that the diagnosis can be made clinically with a high degree of confidence.
9.1.1 Clinical Manifestations
Clinical GBS may be subclassified into several clinicopathological categories (Winer 2011), the most common of which are acute inflammatory demyelinating polyneuropathy (“classic GBS,” AIDP) and acute motor axonal neuropathy (“axonal GBS,” AMAN).
9.1.1.1 Acute Inflammatory Demyelinating Polyneuropathy (AIDP)
Persons of any age may be afflicted with AIDP. In about two-thirds of cases, an antecedent event precedes the illness by 1–3 weeks. Most commonly, this is a nonspecific upper respiratory illness, but organisms associated with AIDP include cytomegalovirus, Epstein–Barr virus, Mycoplasma pneumoniae, and human immunodeficiency virus (HIV). Other possible antecedent events are vaccination, pregnancy, insect bites, and surgery (Arnason and Soliven 1993).
The most important clinical features of GBS (Arnason and Soliven 1993; Ropper et al. 1991 [an exhaustive clinical review], Ropper 1992) are:
1.
Progression to peak deficit over a few hours to 4 weeks, with a monophasic course
2.
Progressive motor and usually sensory deficits in more than one limb
3.
Hypo- or areflexia
Paresthesias often herald the onset, together with or soon followed by weakness. Proximal and distal muscles can be involved, usually symmetrically. The “ascending paralysis” pattern is seen in about half of patients, but upper extremity weakness may predominate from the beginning. Sensory signs are usually overshadowed by motor deficits. Cranial nerve involvement, especially the facial nerve, is common and may be present at the initial presentation. Bulbar and respiratory paralysis requiring intubation occurs in about one-third of patients. Clinical variants of this form of GBS include pure motor or predominantly sensory syndromes, the Miller Fisher syndrome (ataxia, ophthalmoplegia, hyporeflexia), and a dysautonomic picture. Papilledema is seen in up to 5 % of patients.
Elevated CSF protein with a normal cell count (i.e., albuminocytologic dissociation) has historically been very important in delineating GBS but is not seen in as many as 30 % of patients and is often absent in the first weeks of illness (Ropper et al. 1991). Electrophysiological tests are more sensitive, but early in the course, abnormalities may be subtle (Ropper et al. 1991). Typically, there is prolongation of the most proximal and the most distal conduction times, followed by diffuse slowing of conduction velocities, conduction block, and dispersion. Axonal features are frequently found but generally do not predominate.
Cases that progress beyond 8 weeks are best classified as CIDP, and intermediate cases pose a nosologic conundrum. Following maximal deficit, there is usually a stage of stabilization, lasting days to weeks, followed by gradual improvement over weeks to months. The ultimate outcome is good in 80 %, various degrees of deficit remain in 15 %, and death occurs in 5 % of patients (Ropper et al. 1991). Advanced age, a need for mechanical ventilation, and evidence of significant axonal degeneration on electrophysiological tests predict a poor outcome.
Supportive measures, especially for patients requiring respiratory assistance, are the cornerstone of treatment. Plasma exchange (French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome 1987; Guillain-Barre Syndrome Study Group 1985) and intravenous immunoglobulin (van der Meche et al. 1992) benefit many patients, while the consensus is that steroid therapy is of no value (Ropper 1992).
9.1.1.2 “Axonal” GBS (Acute Motor Axonal Neuropathy, AMAN, and Acute Motor Sensory Axonal Neuropathy, AMSAN)
The definition of GBS provided above does not indicate whether an axonal or demyelinating pathology underlies the syndrome. In Europe and the USA, GBS has come to be synonymous with acute inflammatory demyelinating neuropathy (AIDP), its most common form, and axonopathy as a bystander effect (Arnason and Soliven 1993). Some time ago, the existence of a primary “axonal” form was proposed on the basis of a few cases (Feasby et al. 1986). It is characterized by a more aggressive course, poorer outcome, electrical inexcitability and other “axonal” findings on electrophysiological testing, and a stronger association with C. jejuni infection (Yuki et al. 1991; Vriesendorp et al. 1993; Ropper et al. 1991). The existence of this primary axonal process (Feasby et al. 1986) with clinical and electrophysiological features of GBS was not initially accepted as a separate syndrome, and early cases have shown diffuse demyelinating or mixed pathology on autopsy (Fuller et al. 1992; Berciano et al. 1993; Kanda et al. 1989). There was early resistance to the separation of axonal GBS from AIDP since the presence of axonal degeneration in distal nerves is not proof of a primary axonal insult (Feasby et al. 1993). Conversely, distal motor nerve demyelination can account for “axonal” findings on conduction studies (Hall et al. 1992; Triggs et al. 1992) and early studies debated if this entity truly existed or actually represented a GBS variant with primary inflammatory demyelination and superimposed “bystander” axonal damage (vide infra, Cros and Triggs 1994; Fuller et al. 1992; Yokota et al. 1992; Triggs et al. 1992; van der Meche et al. 1991; Brown et al. 1993; Dyck 1993; Feasby et al. 1993; Vallat et al. 1990).
Perhaps the strongest argument for the existence of an “axonal” variant of GBS was provided by the description of Chinese paralytic syndrome, an acute motor neuropathy seen mostly in rural northern Chinese children and having a predilection for the summer season (McKhann et al. 1991, 1993). Autopsy material revealed an axonal noninflammatory neuropathy in some of these patients (McKhann et al. 1993). The observation that 85 % of these patients have serology indicating recent C. jejuni infection (Griffin 1994) has proven of great interest, as the same association was present in some forms of severe GBS and was thought to reflect a common pathogenesis. This pattern of acutely evolving paralytic injury, clinically consistent with GBS, is not confined to China and comprises a significant number of cases of GBS in Asia and Central and South America in which it accounts for 30–65 % of patients. There are clinical differences between AIDP and AMAN, e.g., areflexia is a clinical criterion required to make a diagnosis of GBS, but patients with AMAN have normo- or hyperreflexia. Electrodiagnostic criteria suggest that in Europe and North America, a diagnosis of AMAN was made in only 3–17 % of cases of GBS, whereas the AIDP subtype accounted for 69–90 % (Kuwabara and Yuki 2013).
Currently, AMAN/AMSAN or axonal GBS is accepted as a separate form of GBS different from AIDP (reviewed in Kuwabara and Yuki 2013). AMAN/AMSAN exhibits prominent electrophysiological and pathological evidence of axon loss, with little demyelination, and, frequently, a more aggressive course than AIDP. Antigenic epitopes of infectious agents, such as the lipopolysaccharides of C. jejuni, a Gram-negative poultry-associated organism causing gastroenteritis, are shared with peripheral nerve antigens. The association of C. jejuni with AMAN is more established than with AIDP, and incidence of C. jejuni enteritis may account for incidence variations in forms of GBS in different parts of the world. In addition, population differences in HLA epitopes suggest an immunogenetic component (Ho et al. 1995; Magira et al. 2003).
9.1.2 Pathology
9.1.2.1 General Considerations
Most cases of GBS need not undergo biopsy, as the typical syndrome is easily recognized. The most important alternative peripheral nerve cause of rapidly progressive weakness is vasculitic neuropathy, and there may be times when GBS is so asymmetric or prominently axonal that vasculitis needs to be excluded.
The histological hallmarks of AIDP are a mononuclear inflammatory infiltrate and macrophage-mediated demyelination with a variable amount of axonal damage. Affected nerve may be unremarkable on histological examination (Ropper et al. 1991; Hughes et al. 1992), even when autopsy documents ongoing demyelination elsewhere, usually the nerve root (Kanda et al. 1989; Berciano et al. 1993). Involvement can extend from the spinal root to intramuscular twigs and may result in damage to distal sensory axons and spinal and sympathetic ganglia (Asbury et al. 1969). The sensitivity of peripheral nerve microscopic examination cannot be assessed from the literature because not all patients undergo biopsy; probably more than half of the specimens show specific abnormalities if electron microscopic examination is undertaken (Brechenmacher et al. 1987; Hughes et al. 1992), but a significant minority is normal and another group nonspecific. When sural nerve conduction is normal, biopsy is also likely to be unrevealing (Ropper et al. 1991). In such cases, if it is truly necessary, consideration should be given to biopsy of an electrically involved superficial sensory nerve, or even a motor nerve (Hall et al. 1992). The latter is also a consideration in the uncommon “pure motor” cases of GBS.
9.1.2.2 Light Microscopy
AIDP
An endoneurial mononuclear cell infiltrate composed of macrophages and lymphocytes (chiefly CD4+) is often present (Fig. 9.1), and although the predominant cell type may depend on the stage of evolution of nerve damage, macrophages are usually most numerous (Figs. 9.1, 9.2, and 9.3) (Wisniewski et al. 1969; Brechenmacher et al. 1987; Pollard et al. 1987). The prominence of lymphocytes varies widely in the literature. Asbury and colleagues commented on the “primacy of lymphocytic lesions in idiopathic polyneuritis” (Asbury et al. 1969), but most studies have shown few or no lymphocytes even with immunohistochemistry (Honavar et al. 1991; Mancardi et al. 1988; Hughes et al. 1992; Kanda et al. 1989; Berciano et al. 1993). It is worth noting that only 5 of 57 specimens in the largest GBS nerve biopsy study reported to date demonstrated an increase in endoneurial lymphocytes (Brechenmacher et al. 1987), despite the presence of active demyelination in most of these specimens. It has been argued that the absence of lymphocytes in sural nerve biopsies is due to the limited sampling inherent therein, but several autopsy studies included a close examination of the nerve roots (Honavar et al. 1991; Kanda et al. 1989; Berciano et al. 1993) and did not support this notion. When present, lymphocytes may be perivascular or randomly distributed in the epineurium and endoneurium (Fig. 9.1). Debris-filled macrophages can be seen in association with axons (vide infra), in a perivascular or subperineurial distribution, or randomly dispersed throughout the nerve fascicle (Fig. 9.3). Schwann cells are typically free of myelin debris in AIDP, which differs from typical demyelination in which Schwann cells are the initial degradative site. Schwann cells separate from their myelin sheaths, reenter the cell cycle, produce daughter Schwann cells outside the original basal lamina, and reenter and remyelinate the denuded internode. Axonal loss or axonopathy may represent a “bystander effect” due to endoneurial toxic cytokines (e.g., TNF-α) or edema-induced local ischemia and compression. It is surprising for a demyelinative condition that AIDP often shows considerable loss of axons (Fig. 9.2). Schwann cell proliferation begins at 1–2 weeks after the onset of disease (Asbury et al. 1969) and contributes to endoneurial hypercellularity. Polymorphonuclear infiltrates have been reported in early fulminant disease (Asbury et al. 1969), but we have not observed this in the sural nerve nor has it been noted in other large series (Prineas 1972; Brechenmacher et al. 1987). Plasma cells are infrequently seen. Endoneurial edema is often present but we have never seen it in the absence of other pathology, contrary to the initial reports of Haymaker and Kernohan (1949). Months and even years after the acute illness, inflammatory cells may still be seen in peripheral nerve (Asbury et al. 1969).
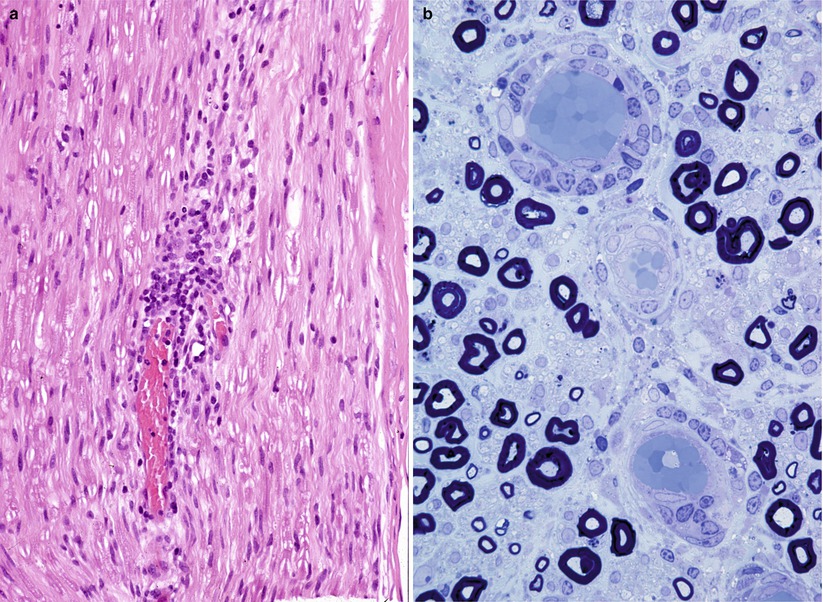
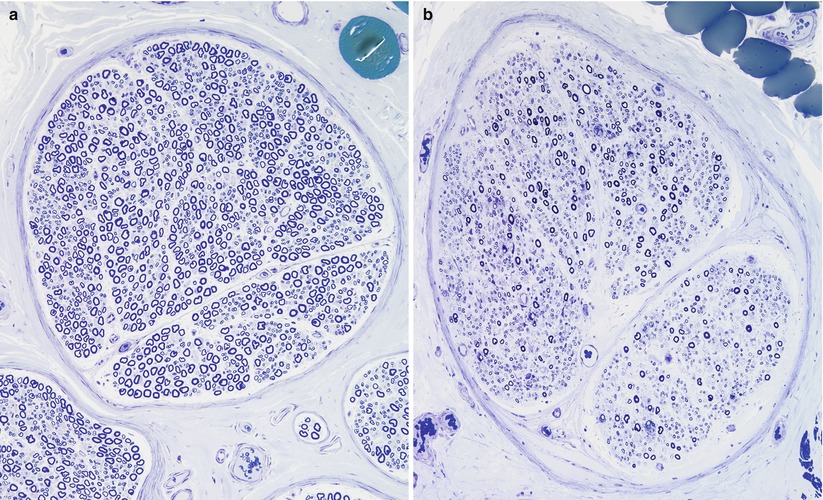
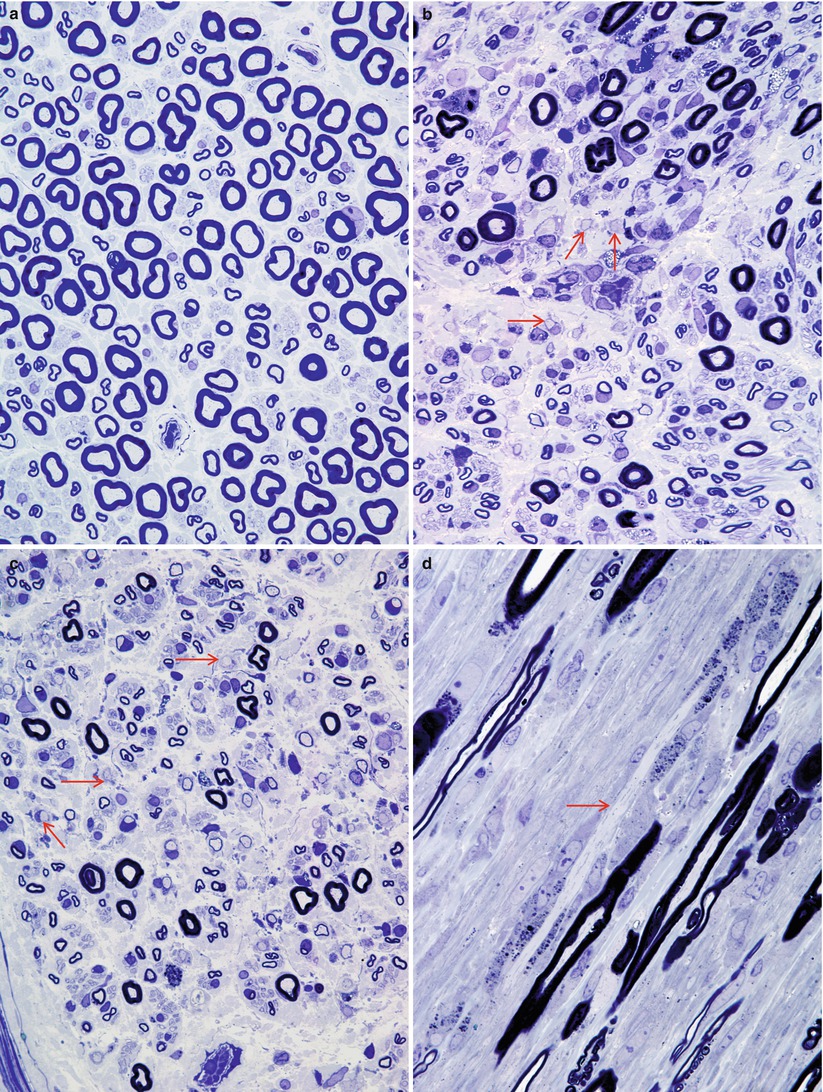
Fig. 9.1
GBS: note endoneurial perivascular cuffing with mononuclear cells in H&E and 1 µ plastic sections (a, paraffin 400×; b, plastic 1,000×)
Fig. 9.2
Fiber density and endoneurial cellularity are contrasted in normal (a) and GBS, AIDP subtype (b) sural nerves (a, b: 1 µ plastic sections, 200×)
Fig. 9.3
GBS, AIDP subtype: normal sural nerve (a) compared with the typical appearance of Guillain–Barré syndrome (b–d). Note the large number of elongated and irregular cells in the endoneurium, loss of MF number, and myelin breakdown with numerous denuded axons (arrows, b, c). (d) Longitudinal section shows a naked axon (arrow) surrounded by multiple Schwann cells and macrophages with myelin debris (a, b, 1 µ plastic section 600×; c, d 1 µ plastic section 1,000×)
The predominant pathological process in AIDP is segmental demyelination and remyelination. The entire spectrum of normally myelinated, thinly myelinated, and denuded axons can be seen (Figs. 9.2 and 9.3). A longitudinal section shows a long expanse of naked axon (arrow, Fig. 9.3d) surrounded by multitudes of closely applied, myelin-containing macrophages. The severity of these changes often depends on the duration of the disease. Some cases may show selective involvement of large myelinated fibers. Active axonal degeneration is not uncommon, but this is usually minor relative to the demyelinating changes. A demyelinated axon with an adjacent macrophage filled with myelin debris may be mistaken for a myelin ovoid of axonal degeneration if the attenuated axon is not visualized by light microscopy. As Schwann cells do not accumulate large amount of debris, any cell laden with myelin breakdown products is likely to be a macrophage. Axonal degeneration should not dominate the picture if a diagnosis of “primary demyelination consistent with GBS” is to be made (however, exceptions exist, vide infra).
Widening of the nodal gap, best seen on teased fiber preparations, is an early but nonspecific alteration, whether in GBS (Asbury et al. 1969) or in other demyelinating or axonal neuropathies. Onion bulbs have been seen in cases reported as GBS (Prineas 1972), but their presence suggests chronicity and that the patient more likely suffers from CIDP presenting with an acute exacerbation of previously subclinical disease.
AMAN/AMSAN
In the initial case of axonal GBS (AMAN) studied at autopsy, peripheral nerve showed only axonal degeneration and scanty inflammation (Feasby et al. 1986) which overlaps with some cases of clinically typical GBS (Prineas 1972; Hughes et al. 1992; Vallat et al. 1990). Although typical cases of AMAN show mild sensory axon degeneration, some cases of axonal GBS may show extensive sensory changes which are designated acute motor sensory axonal neuropathy (AMSAN). These cases are thought to represent a spectrum of a single type of immune attack on the axon differing in severity, immunologic presentation, or distribution of a critical epitope (Griffin et al. 1996, vide infra). Since the recognition of additional cases of AMAN/AMSAN, it has become clear that the most striking differences between AIDP and AMAN/AMSAN are the paucity of lymphocytic inflammation and the lack of demyelination and a variable increase in the number of degenerating axons in AMAN/AMSAN. Axonopathy ranges from diffuse axonal degeneration to distal axonal damage, with denervated neuromuscular junctions; the latter changes may be reversed rapidly.
Case 9.1
We have seen an illustrative case of AMAN of a 66-year-old Japanese woman who experienced a flu-like illness approximately 3 weeks prior to the initial onset of paresthesias of distal hand and feet with weakness which soon dominated her relatively minor sensory symptoms. During the course of a single day, she rapidly progressed to a condition where she could not stand without support and had difficulties swallowing and speaking, resulting in hospitalization. At that time, she was awake, alert, and fully oriented with normal pupillary light reflex and extraocular movement but mild weakness of facial muscles and tongue protrusion. Her arms and legs were weak bilaterally, and tendon reflexes were diminished or absent throughout. On the next hospital day, she was completely tetraplegic with absent tendon reflexes. On the third hospital day, she suddenly developed circulatory collapse, was resuscitated, but remained in persistent coma thereafter. The patient died with a total duration of illness of 41 days. Autopsy was performed and her anterior and posterior roots were sampled extensively.
Her lumbar anterior roots were most heavily involved. The proximal portion of her L4 anterior root showed occasional degenerating axons (Fig. 9.4a), accompanied by little inflammatory or macrophage infiltration. The distal portion of the same L4 anterior root, however, showed extensive axonal degeneration (Fig. 9.4b) with macrophage infiltration. Another portion of L4 anterior root showed residual degenerating axons but well-established regenerating axonal clusters (arrows, Fig. 9.4c), resembling the minifascicles of a neuroma. Sampled thoracic anterior roots also show ongoing axonal degeneration, although at that level, dorsal roots appeared minimally involved (Fig. 9.4d). Macrophages are found adjacent to the axons, in an expanded node of Ranvier (Fig. 9.5a) or within intact myelin sheaths (Fig. 9.5b, c) (case provided by Drs. Shinji Ohara, Mitsunori Yamada, and Hitoshi Takahashi).
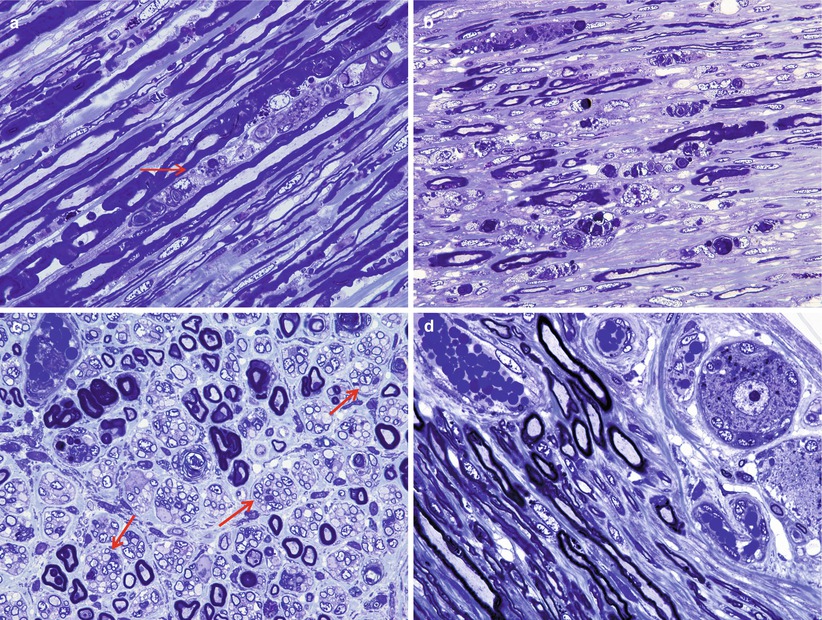
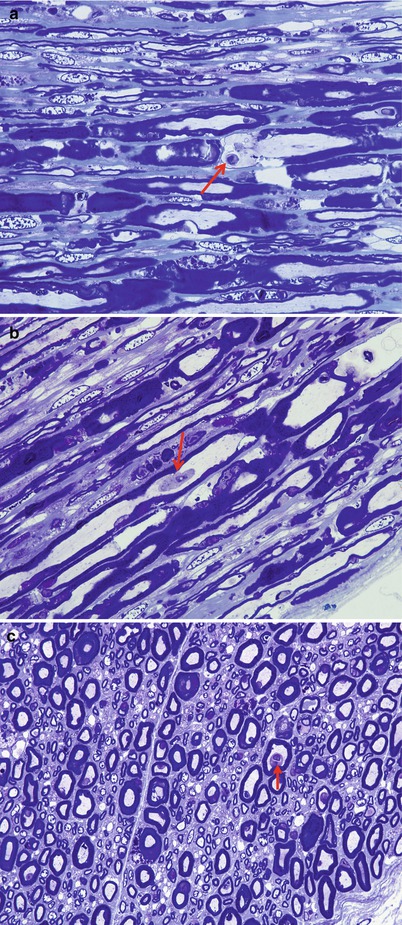
Fig. 9.4
GBS, AMAN subtype: lumbar (L4) proximal anterior roots showing individual axonal degeneration (arrow, a) or, in its more distal segments, extensive degeneration (b). (c) Rare degenerated axon surrounded by numerous clusters of regenerating axons (arrows). (d) Proximal segment of thoracic posterior root shows a DRG neuron and no axonal degeneration (1 µ thick plastic sections; magnification: a–d, 400×) (Case provided by Drs. Shinji Ohara, Mitsunori Yamada, and Hitoshi Takahashi)
Fig. 9.5
GBS, AMAN subtype: L4 anterior roots show paranodal swelling with infiltrating macrophages (arrow, a). Longitudinal and cross sections show macrophages (arrows, b, c) within intact myelin sheaths adjacent to intact axons (1 µ thick plastic sections; magnification: a–c, 1,000×)
9.1.2.3 Electron Microscopy
AIDP
Ultrastructural examination shows a complex collection of pathological findings (Fig. 9.6) including naked axons, edema separating axons, a background of macrophages with debris, demyelination (arrowhead, Fig. 9.6), and remyelination (arrows, Fig. 9.6). Ultrastructural analysis is critical for establishing the diagnosis of macrophage-mediated demyelination (Figs. 9.7, 9.8, and 9.9), the hallmark of inflammatory demyelinating neuropathy, whether acute (GBS) or chronic (CIDP). The classic picture is of a macrophage penetrating the Schwann cell basement membrane (Fig. 9.7a, b, arrows), separating Schwann cell cytoplasm from its underlying intact myelin (Fig. 9.7c) and insinuating cytoplasmic fingers into the intraperiod lines of the myelin sheath. Myelin destruction may seem to occur by dissolution along the site of contact with macrophage processes or through the separation, engulfment, and removal of large segments of myelin off the axon. The invading macrophage is rich in organelles including smooth ER, mitochondria, Golgi, and ribosomes, but the processes inserted into the myelin lamellae are largely free of these. All the while, the axon maintains a relatively normal appearance. Sometimes several processes may attack the myelin sheath at once, and it is unclear if they belong to one or several macrophages. Debris may appear as pieces of normally compacted myelin, vesicular myelin, lamellar disorganized osmiophilic material, or amorphous globules (Figs. 9.8, 9.9, and 9.10). Rarely, debris that seems to be extracellular may be detected (Prineas 1972).
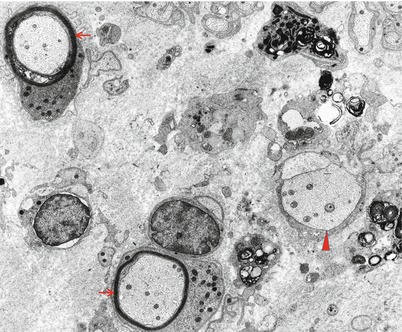
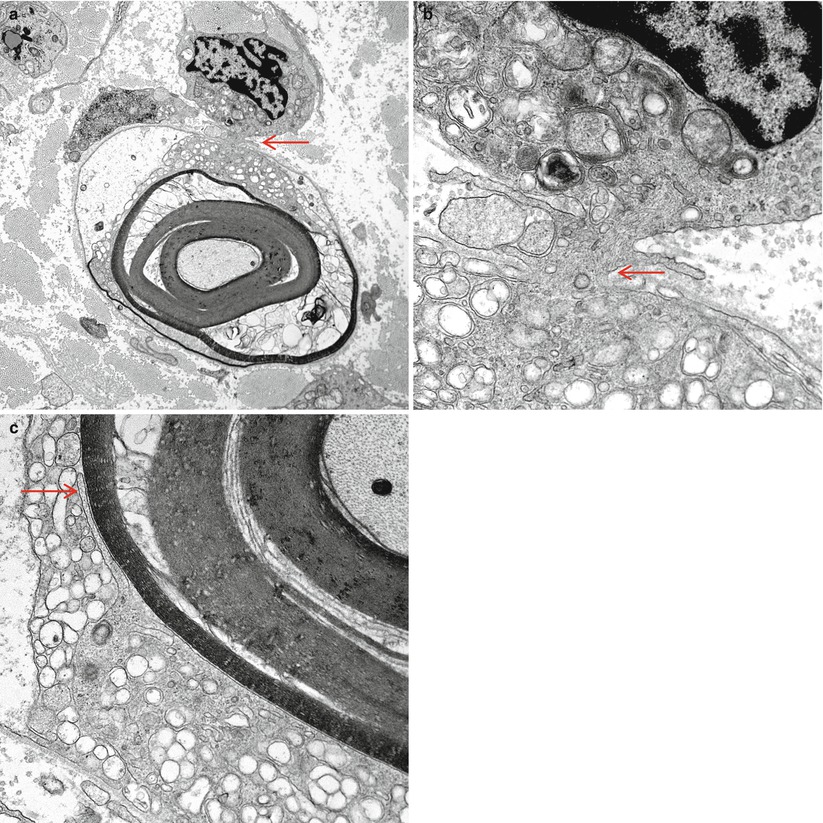
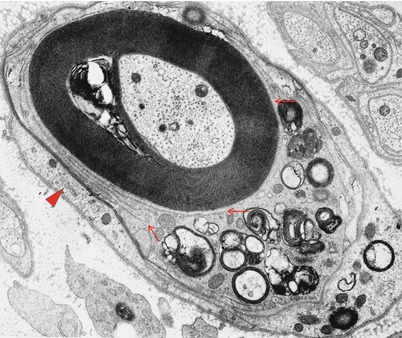
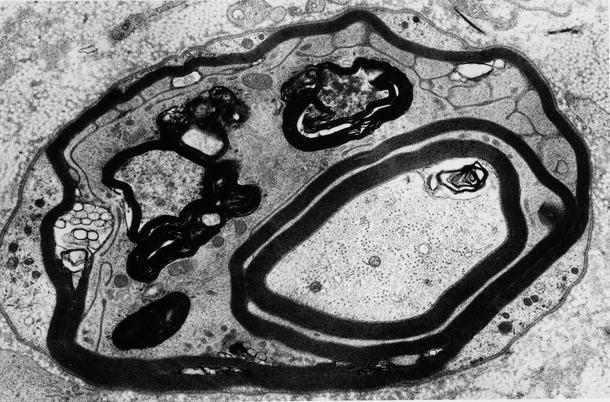
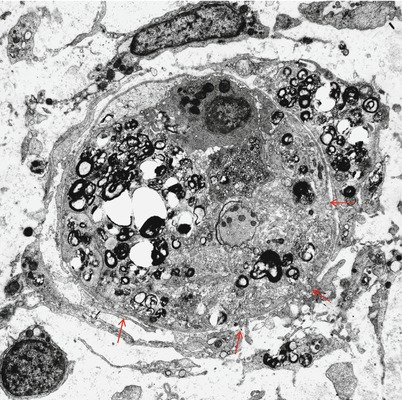
Fig. 9.6
GBS, AIDP subtype at 5 weeks: the endoneurium contains macrophage processes laden with myelin debris, a demyelinated axon (arrowhead), and two remyelinating fibers (arrows) (×9,256, from Bilbao 1995)
Fig. 9.7
GBS, AIDP subtype: a macrophage has begun to insinuate processes into the Schwann cell/axon complex at the arrow (a), seen at higher magnification in (b). The myelin has begun to disintegrate in response to the macrophage which contains relatively little digested myelin. At the site indicated (arrow, c), higher magnification shows a macrophage process adjacent to the outermost myelin lamella (Magnification: a, 6,000×; b, 40,000; c, 25,000×)
Fig. 9.8
GBS, AIDP subtype: an intratubal macrophage process strips myelin. Note interleaving of myelin lamellae and tongue-like macrophage extensions (arrows). The Schwann cell has been pushed peripherally (arrowhead) (Magnification: 20,800×)
Fig. 9.9
GBS, AIDP subtype: intramyelinic macrophage process displays mitochondria and vacuoles containing myelin fragments. Adjacent myelin is being “peeled off” (15,620×)
Fig. 9.10
GBS, AIDP subtype: advanced stage of demyelination in a fiber whose shrunken axon is at the center. The Schwann cell basal lamina is penetrated in at least four sites (arrows). Macrophage processes and a lymphocyte are seen outside the fiber (8,914×, from Bilbao 1995)
The picture described above is not always easy to demonstrate. A more common observation is the presence of supernumerary cell processes sharing the Schwann tube with a residual Schwann cell and a demyelinated axon (Fig. 9.8); further afield, a macrophage may be present but separated from the affected fiber. Presumably the site of macrophage penetration has occurred outside the plane of section. As Schwann cells do not commonly accumulate myelin debris in AIDP, one may infer that the cell processes harboring phagocytosed lipid debris are those of a macrophage (Prineas 1972). The Schwann cell may be absent altogether, with the axon sharing a Schwann tube only with macrophages. Demonstration of segmental demyelination alone cannot establish macrophage-mediated demyelination. In summary, what is characteristic about macrophage-mediated demyelination is separation or peeling off of normal appearing myelin by fingerlike macrophage extensions; this process cannot be visualized after myelin disintegration is complete, and in such a situation, it cannot be determined whether the macrophage is the primary effector of the damage or simply a scavenger cell (Fig. 9.10). Alterations of myelin periodicity have very rarely been reported in GBS, including both uncompacted myelin (Brechenmacher et al. 1987) and widely separated myelin involving the two or three outermost turns of the myelin sheath (Vallat et al. 1994).
Schwann cells can appear normal as they are being separated from their myelin or may show reactive or degenerative changes, with dilated ER, loss of organelles, and watery distension. Little or no myelin debris accumulates within them. Most axons appear unperturbed in the face of the myelin destruction surrounding them, but some show various stages of degeneration, ranging from loss of electron density to Wallerian degeneration. Naked axons have been said to shrink by as much as 50 % in volume as a consequence of the myelin loss, with a resulting increased density of tubules and filaments, and wrinkling of the axolemma (Prineas and McLeod 1976; Carpenter 1972; Raine et al. 1969). Evidence of recent remyelination is found in the presence of axons surrounded by very thin, poorly compacted myelin sheaths and Schwann cells rich in organelles.
A loss of unmyelinated axons may be seen but is rarely prominent and never predominant (Prineas 1972). Tubuloreticular inclusions have rarely been seen in endothelial cells and macrophages in GBS associated with HIV infection (Fuller and Jacobs 1989) but never in classic GBS. On two occasions, we have seen mononuclear cells, free of debris, within a seemingly intact myelin sheath without any detectable axon. These were cases with a particularly aggressive course and prominent axonal degeneration, but the observation has been too infrequent to allow generalization. We have never observed this finding outside of GBS. Similar observations were reported by Brechenmacher et al. (1981). A single report claims to document “viral particles” within the nerve in classic GBS (Sibley 1972), but in the absence of any corroborating observations, this must be regarded with skepticism. PCR studies have failed to demonstrate evidence of CMV genome in nerve specimens of patients with GBS (Hughes et al. 1992).
Although myelinated axon numbers are clearly decreased in GBS (Fig. 9.2b), ongoing axonal degeneration may be infrequent at any one time. Nonetheless, some cases of GBS exhibit so much axonal degeneration (Fig. 9.11a) that they can be mistaken for pure axonal degeneration. The case illustrated in Fig. 9.11 was initially thought to represent fulminant axonal degeneration alone; however, examination of other fascicles where myelinated axons were still present demonstrated macrophage-mediated demyelination (Fig. 9.11b, c).
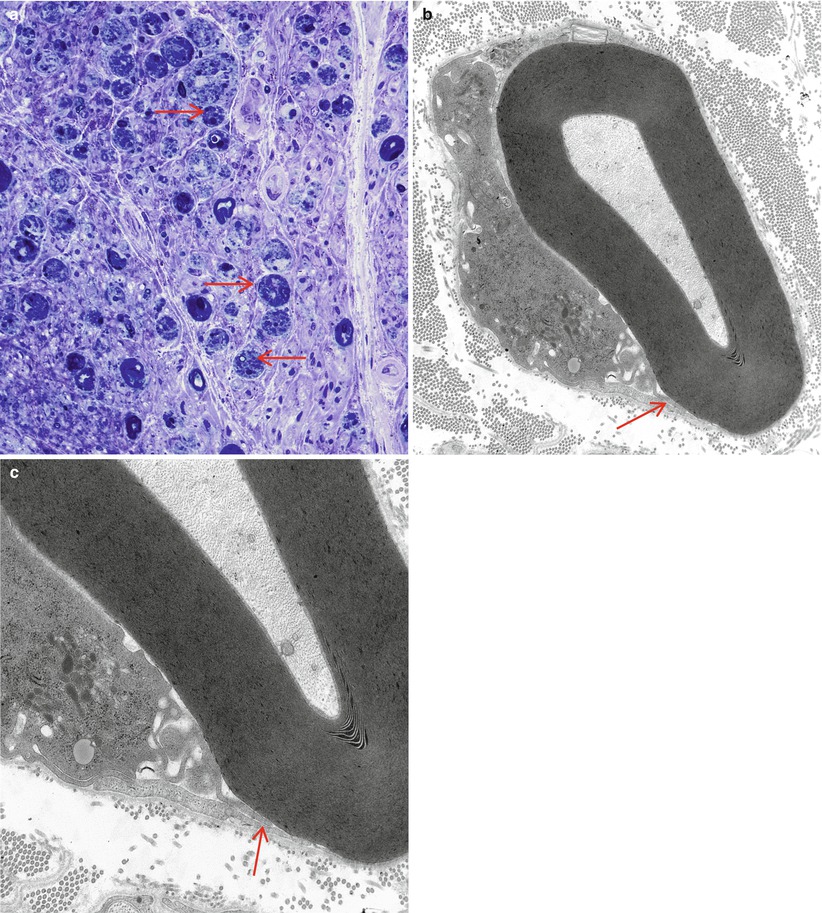
Fig. 9.11
GBS, AIDP subtype, with marked axonal degeneration. Numerous actively degenerating axons (arrows, a) do not suggest a demyelinative process. Nonetheless, at other sites in an adjacent fascicle, the characteristic macrophage-mediated demyelination (arrows, b, c) identifies this as a case of GBS, AIDP subtype (a, 1 µ plastic section, 1,000×; b, 12,000×; c, 20,000)
AMAN/AMSAN
Ultrastructural changes include penetration of macrophage processes into the periaxonal space at the paranode or more distantly (Fig. 9.12a) and demonstration of an entire macrophage within myelin sheaths adjacent to intact axons (Fig. 9.12b, different case). Altered nodal structure with widening of the nodes and distortion of paranodal myelin may be followed by degeneration of outermost myelin terminal loops, eventually causing local degeneration of Schwann cell cytoplasm but sparing the internodal myelin sheath.
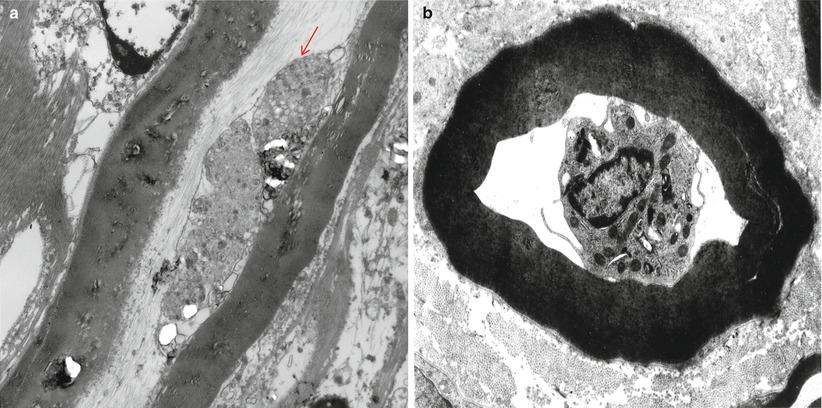
Fig. 9.12
GBS, (axonal type, AMAN): (a) a macrophage process (arrow) adjacent to an intact axon and within an intact myelin sheath. (b) A mononuclear cell, most likely a macrophage, surrounded by an intact myelin sheath (different case) (Magnification: a, 12,000×; b, 4,560×)
9.1.2.4 Immunohistochemistry
As in other inflammatory neuropathies, the use of immunohistochemical stains for leukocytes may bring out subtle infiltrates. Increased cellularity of the endoneurium can be due to macrophages, Schwann cells, or lymphocytes which can be separated by immunohistochemistry. However, the range of normal is not well established, and anything less than perivascular cuffing is of uncertain significance, especially in the epineurium.
An immunohistochemical and immunofluorescence search for IgG, IgM, or complement deposition on Schwann cells and myelin is usually (Brechenmacher et al. 1987; Hughes et al. 1992; Honavar et al. 1991) but not always (Luijten and Baart de la Faille-Kuyper 1972; Nyland et al. 1981; Vallat et al. 1994) negative in AIDP. Immunohistochemistry may be useful in characterizing the binding of anti-ganglioside antibody at the node in AMAN/AMSAN.
9.1.3 Pathogenesis
In the AIDP form of GBS, the fundamental process is primary demyelination, and axonal degeneration is variable in severity. Pathological alterations occur throughout the peripheral nervous system, not necessarily most prominently at the spinal nerve roots (Asbury et al. 1969). The experimental literature pertaining to pathogenesis has focused at various times on lymphocytes, macrophages, humoral factors, and alterations in the blood–nerve barrier. In AMAN, the primary target of the immune attack is the axon (particularly nodal/paranodal gangliosides) rather than the Schwann cell or myelin, with a predilection for motor axons. Recent evidence suggests that the node of Ranvier is the primary target of immune attack in patients with both forms of GBS and CIDP (Devaux 2012), an interpretation which has been proposed and supported by skin biopsies (Doppler et al. 2013) using immunolocalization of the proteins that characterize the node (voltage-gated sodium channels) and paranodal region (neurofascin).
9.1.3.1 Lymphocytes
Emphasis on the role of lymphocytes began largely with an extensive autopsy review in 1969 which showed many of these cells in peripheral nerve at the earliest stages of GBS (Asbury et al. 1969). The importance of lymphocytes in pathogenesis has subsequently been supported by studies of animal models for GBS (vide infra). Interestingly, authors of autopsy and sural nerve biopsy reports have commented on the relative paucity of lymphocytes in their material (Honavar et al. 1991; Brechenmacher et al. 1987; Mancardi et al. 1988; Kanda et al. 1989). This may indicate that more than one pathophysiological mechanism may cause the same clinical picture or, more likely, that the disease is being examined at different stages.
The experimental allergic neuritis (EAN) animal model, in its numerous permutations, has been the cornerstone of research in GBS (Waksman and Adams 1955; Raine 1985; Hartung and Toyka 1990; Hahn et al. 1990). Experiments suggest that EAN is a T-cell-mediated autoimmune disease (Hartung and Toyka 1990). Starting 8–12 days after injection, lymphocytes and macrophages migrate across endothelial capillaries into the interstitium (Hahn et al. 1985), and demyelination soon follows. The most important antigen inciting EAN appears to be P2 protein (Rostami et al. 1984). CD4+ T cells sensitized to whole myelin, P2 protein, and even certain neuritogenic fragments of P2 protein can transfer the disease (Linington et al. 1984; Rostami et al. 1985; Rostami 1993). The severity of EAN is markedly diminished in T-cell-depleted recipient animals (Brosnan et al. 1987, 1988; Strigard et al. 1988). Administration of antibody to interleukin-2 receptor blocks activation of T cells and prevents development of the adoptive disease (Hartung et al. 1989).
In human material, some authors have emphasized infiltration with lymphocytes as the earliest change (Asbury et al. 1969; Iqbal et al. 1981). Serum IL-2 and its receptor are increased in patients with GBS relative to controls (Hartung and Toyka 1990) and correlate with disease activity (Hartung et al. 1991). Detection of specific sensitization of GBS patients’ lymphocytes to P2 protein has been inconsistent (Taylor et al. 1991; Sheremata et al. 1975; Iqbal et al. 1981; Burns et al. 1986), and treatment with anti-lymphocyte antibodies has had no effect on the disease (Feasby 1991).
The pathogenesis of AMAN may initially involve antibody targeting of a constituent of the node of Ranvier or paranodal axolemma, most likely a ganglioside, to which it binds, fixes complement, and secondarily recruits macrophages; these events are thought to culminate in axonal dysfunction or, in some cases, axonal degeneration. In support of this hypothesis, rabbits immunized with gangliosides or GM1 may develop weakness, high titers of anti-GM1 antibodies, and pathological changes similar to human AMAN, with axonal degeneration confined to motor roots largely in the absence of inflammation (Susuki et al. 2003, 2007). Some people immunized with gangliosides have developed axonal forms of GBS.
9.1.3.2 Macrophages
Macrophages play a central role in inflammatory demyelination. At the height of disease activity, they form the predominant inflammatory cell (Iqbal et al. 1981; Brechenmacher et al. 1987). Macrophages are the dominant infiltrating cell type after the earliest stages of EAN (Lampert 1969; Raine 1985), and suppression of their activity diminishes severity of demyelination (Hartung et al. 1988). Ultrastructural studies in both human and animal inflammatory demyelination demonstrate that myelin destruction begins when a macrophage inserts a process through the basement membrane surrounding a myelinated fiber. Myelin lamellae are split, and resulting debris is carted away in the macrophage. In human disease, macrophage-mediated myelin stripping is only seen in CIDP and GBS. Reasons for the specific attack on myelin remain unclear. Complement and immunoglobulin deposits have been described on Schwann cells in AIDP (Luijten and Baart de la Faille-Kuyper 1972; Nyland et al. 1981) and may serve to opsonize the myelin sheath and promote macrophage adhesion and attack. However, this finding is inconsistent (Brechenmacher et al. 1987; Hughes et al. 1992) and nonspecific (Hays et al. 1988).
Macrophages may also play an important role in axonal damage. It has been noted that in both EAN and human inflammatory demyelinating neuropathies, the severity of axon loss is correlated with the severity of local inflammation and demyelination (Asbury et al. 1969; Feasby et al. 1990; Madrid and Wisniewski 1977). Tumor necrosis factor, a cytokine released by activated macrophages, has been shown to cause axonal degeneration (Said and Hontebeyrie-Joskowicz 1992). Macrophages are directly related to the degree of axonopathy in AMAN/AMSAN in response to periaxonal invasion.
9.1.3.3 Antibodies and Complement
In patients with GBS, circulating antibodies to peripheral nerve tissue are found in higher titers than in noninflammatory neuropathies or controls (Lundkvist et al. 1989; van Doorn et al. 1987). Complement-fixing antibody to peripheral nerve myelin is present in the serum of up to 95 % of GBS patients (Koski 1990). Antibody titers may correlate with clinical (Vriesendorp et al. 1991; Koski et al. 1986) or histological (Koski 1990) disease severity or response to treatment (van Doorn et al. 1987). However, some observers have failed to detect anti-myelin antibodies (Winer et al. 1988), and while IgG can pass the placental barrier from mother to fetus, maternal GBS does not cause neuropathy in the neonate (Ropper et al. 1991). Moreover, the proven efficacy of plasma exchange may result from removal of circulating antibodies, but an alternative explanation is the removal of cytokines (Pollard 1987) or a circulating antigen (Harvey et al. 1988).
Immunoglobulins may mediate myelin damage through activation of complement. Serum from Guillain–Barré AIDP patients injected into neural cultures and animal nerve may cause demyelination (Saida et al. 1982; Feasby et al. 1982; Sawant-Mane et al. 1991), even in the absence of macrophages (Birchem et al. 1987). Terminal complement complex (Koski 1990) is required for this demyelination to take place (Sawant-Mane et al. 1991; Feasby et al. 1987). The complement cascade terminates with formation of C5b-9 membrane attack complex, a transmembrane channel which causes cell lysis by allowing ionic influx. The resulting influx of calcium may activate endogenous neutral proteases which can cleave important myelin proteins. Indeed, calcium influx into Schwann cells results in vesicular demyelination and macrophage-mediated myelin stripping (Smith and Hall 1988). In vivo, markers of complement activation are present both in serum and on spinal nerve roots and peripheral nerves of patients with GBS (Koski et al. 1987; Sanders et al. 1986).
Immunoglobulins may mediate myelin damage through activation of macrophages.
Transient complement deposits on Schwann cell and myelin membrane were seen in peripheral nerve in an EAN model before the onset of disease, and regions of accumulations of complement correlated with sites of macrophage concentration (Stoll et al. 1991). In experimental allergic encephalomyelitis, myelin lamellae are attached to coated pits on the macrophage surface, suggesting receptor–ligand interaction (Epstein et al. 1983). However, the finding of immunoglobulin or complement deposits on myelin or Schwann cell membranes is inconsistent (Luijten and Baart de la Faille-Kuyper 1972; Nyland et al. 1981; Brechenmacher et al. 1987; Hughes et al. 1992; Schenone et al. 1988).
The antigenic specificity of the putative complement-fixing or macrophage-activating anti-myelin antibodies is an area of active research. Several myelin antigens have been implicated including GM1 and a number of neutral and acidic myelin glycolipids. Some of these myelin antigens have cross-reactivity with antigens on organisms which have been epidemiologically associated with GBS: GM1 with Campylobacter jejuni and neutral myelin glycolipids with Forssman antigen, seen on a variety of viral agents (Koski 1992). However, these antibodies have been seen in a low percentage of patients with inflammatory demyelinating polyneuropathies and in a high proportion of controls, and there may be no association between the organism and its alleged cross-reactive peripheral nerve antigen (Quarles et al. 1990; Svennerholm and Fredman 1990; Vriesendorp et al. 1993; Enders et al. 1993; Mithen et al. 1992).
AMAN is associated with serum IgG binding to GM1, GD1a, and GalNAc-GD1a gangliosides. The selectivity of AMAN for motor axons may reflect the different affinities of anti-GD1a antibodies for sensory GD1a and motor GD1a with some evidence for preferential staining of motor axons and a subpopulation of small sensory axons (Lunn et al. 2000). Immunoglobulin G (IgG) anti-GD1a antibodies were found in 60 % of Chinese patients with axonal GBS but in only 4 % of patients with AIDP (Lunn et al. 2000), whereas anti-GM1 antibodies were frequent in both variants (Ho et al. 1999). Recently, AMAN and AMSAN with their strong association with antibodies to gangliosides have been classified as nodo-paranodopathy (Uncini et al. 2013). Others have proposed that autoantibodies targeting antigens in the presynaptic membrane of the NMJ (Fewou et al. 2013) represent an additional selective mode of access. Evidence is emerging to show that at least some antibodies (“ganglioside complexes antibodies”) can induce pathogenic effects (Rinaldi 2013). Although antibodies against gangliosides GM1, GM2, basal lamina components, and several myelin proteins have been identified in some cases (Griffin and Sheikh 2005), no characteristic pattern of anti-ganglioside antibodies has been established in AIDP (compared with acute motor axonal neuropathy, AMAN, vide infra). Different serotypes of ganglioside-like polysaccharide antigens in C. jejuni may influence the likelihood of AMAN since only a small minority of patients with C. jejuni enteritis develop AMAN. Both AMAN and AMSAN are thought to be a part of the spectrum of a single type of immune attack on the axon (Griffin et al. 1996), a contention supported by the presence in both entities of anti-GM1 (64 %), anti-GM1b (66 %), and anti-GalNAc-GD1a (33 %) IgG antibodies (Yuki et al. 1999; Hughes and Cornblath 2005) and the association of both conditions with C. jejuni enteritis.
Experimental animal studies have shown immunization with a bovine brain ganglioside mixture, or isolated GM1 produces antibody deposition at nodes, monophasic weakness, and prominent axonal degeneration without lymphocytic infiltration or demyelination. The final product of complement activation, the complement membrane attack complex, is formed at the nodal axolemma. Nodal sodium channel clusters disappear as complement is deposited and the node becomes exposed to the paranodal regions, lowering the safety factor. Autoimmune attack causes the disappearance of adhesion molecules, such as contactin and contactin-associated protein, at the paranodes, and disrupts the axoglial junctions and detachment of paranodal myelin terminal loops, as seen with electron microscopy (Kuwabara and Yuki 2013).
9.1.3.4 Alteration in the Blood–Nerve Barrier
Serum from GBS patients has no demyelinating activity unless it somehow bypasses the blood–nerve barrier (Tandon et al. 1980). In the EAN model, vascular permeability changes coincident with immunoglobulin changes have been observed before or along with other signs of pathology (Powell et al. 1983; Hahn et al. 1985). One group working with the EAN model demonstrated an increase in mast cell degranulation before the appearance of lymphocytes and onset of symptoms and showed that antagonism of mast cell vasoactive amine activity at the right time delayed the onset of EAN symptoms (Brosnan et al. 1990). Thus, early permeability changes in the blood–nerve barrier may play an important role in the pathogenesis of GBS.
9.1.3.5 Summary
Influenced by analysis of the experimental allergic neuritis, AIDP T cells were thought to recognize an autoantigen, perhaps an epitope on an infectious agent (“molecular mimicry”), in the presence of MHC class II and co-stimulatory molecules on the surface of an antigen-presenting cell. Nonetheless, no characteristic pattern of induced antibodies has been identified in AIDP as it has in AMAN.
9.1.4 Differential Diagnosis
Diseases of muscle, nerve root, and spinal cord that superficially resemble GBS are sorted out on clinical grounds. Common in clinical neuropathology are nonspecific findings in a nerve biopsy such as chronic inflammation and axonal degeneration, both being features of GBS and vasculitic neuropathy. Epineurial inflammation, presence of granulocytes, and intense and multifocal axonal damage favor vasculitis, whereas endoneurial inflammation, macrophage infiltration, and milder axonal degeneration argue for GBS.
Nonspecific histological findings that are not out of keeping with GBS can be seen in other neuropathies included in the clinical differential diagnosis. Such neuropathies include paraneoplastic sensorimotor neuropathy, paraprotein-associated neuropathy (Chap. 14), porphyria, Lyme neuritis, alcohol (Tabaraud et al. 1990), arsenic and thallium poisoning (Chap. 18), and neurolymphomatosis (Diaz-Arrastia et al. 1992). We have also seen a patient with granulomatous polyneuropathy evolving at the tempo of GBS (Chap. 10, case 10.1).
Macrophage-mediated demyelination is the only pathognomonic feature of GBS, AIDP subtype (or CIDP). In the search for macrophage penetration of Schwann cells and myelin, we resort to surveying many semithin sections under oil immersion and then selecting suggestive areas for ultrastructural examination. Vesicular degeneration of myelin is less specific. The finding of onion bulbs in the case of putative GBS suggests CIDP.
9.2 Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Patients suffering from what is now called chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) have been described under several names in the past, among them chronic relapsing polyneuritis (Prineas and McLeod 1976), hypertrophic interstitial radiculoneuropathy (Austin 1956), chronic relapsing dysimmune polyneuropathy (Dalakas and Engel 1981), and chronic Guillain–Barré syndrome (Thomas et al. 1969). The clinical and pathological spectrum has been clearly delineated (Dyck et al. 1975; Prineas and McLeod 1976; McCombe et al. 1987; Barohn et al. 1989), and with the appreciation that many of these patients are responsive to therapy and that this is a common peripheral neuropathy (Dyck et al. 1982a), CIDP has become a very important neurological syndrome.
9.2.1 Clinical Manifestations
9.2.1.1 The Typical Syndrome
Criteria for the diagnosis of CIDP have been formulated by several groups (Barohn et al. 1989; Dyck et al. 1975, 1993; Ad Hoc Subcommittee 1991). Clinical features include:
1.
Evolution over at least 2 months, often over many months
2.
Progressive or relapsing and remitting course
3.
Motor and/or sensory (usually both) symptoms and signs
4.
Hypo- or areflexia
Onset may be at any age, and involvement is usually symmetrical. Motor manifestations tend to predominate, with proximal and distal muscles being involved. Large fiber sensory deficits can be prominent (vibration, joint position), but usually all modalities are affected. Cranial nerve palsies, frequently cranial nerve VII, occur in 14–43 % of patients (Barohn et al. 1989; Prineas and McLeod 1976; McCombe et al. 1987), and autonomic dysfunction is an uncommon feature. A palpably enlarged nerve is found in 11 % (Dyck et al. 1975; Prineas and McLeod 1976) and unexplained papilledema in as many as 7 % (Dyck et al. 1975).
CSF protein is elevated and cell count is normal in 90–95 % of patients (Dyck et al. 1975; Prineas and McLeod 1976; Barohn et al. 1989). Nerve conductions usually give evidence of a demyelinating process which is most severe proximally, but a variable amount of axonal disease can be seen (Prineas and McLeod 1976; Dyck et al. 1975; McCombe et al. 1987; Barohn et al. 1989). Most patients show conduction slowing in at least two peripheral nerves. Sensory potentials may be lost, and motor distal latencies become prolonged. Because the brunt of the disease falls proximally and standard nerve conduction studies assess the distal nerve better than the proximal nerve, abnormalities may be subtle early in the course. EMG examination may show acute or chronic neurogenic changes.
9.2.1.2 Diseases Associated with CIDP
A circulating paraprotein is seen in 8–30 % of cases (Barohn et al. 1989; Bromberg et al. 1992), but some authors exclude these patients (Dalakas and Engel 1981; Dyck et al. 1975) or classify them as having CIDP associated with a concurrent disease (Ad Hoc Subcommittee 1991). Clinically, there is no major difference between the two (Bromberg et al. 1992), although IgM paraprotein-associated neuropathies may enjoy a special status (Chap. 14). Classically, the pathology of paraproteinemic neuropathy and CIDP differs, but there can be an overlap. It remains to be determined whether CIDP with or without a paraprotein are different pathophysiological entities (Dyck 1990). Similar considerations apply to HIV-positive patients who can develop a CIDP-like syndrome with identical clinical, pathological (Vital et al. 1992), and therapeutic response patterns to those seen with classic CIDP (Cornblath et al. 1987), except for the tendency for CSF pleocytosis associated with HIV infection. Furthermore, a CIDP-like picture has infrequently been described in patients with SLE, chronic active hepatitis, ulcerative colitis, lymphoma (Sumi et al. 1983), and other systemic disease including diabetes, malignancy (e.g., melanoma), Sjögren’s syndrome, and hepatitis C, and it is unclear whether there is any distinction between such cases and isolated CIDP (Rechthand et al. 1984; Barohn et al. 1989). In diabetic individuals, CIDP is reported to be more severe, but this is not established.
The distinction between recurrent Guillain–Barré syndrome (GBS) and relapsing CIDP may be difficult (Grand’Maison et al. 1992). Complete recovery between attacks favors the former (Dyck et al. 1993). The relationship between CIDP and multiple sclerosis is tantalizing, as central nervous system white matter involvement is known to occur in cases which are otherwise typical of CIDP (Dyck et al. 1975; Mendell et al. 1987; Uncini et al. 1991; Thomas et al. 1986), and peripheral nerve dysmyelination has been documented in multiple sclerosis (Schoene et al. 1977; Pollock et al. 1977). Whether any or all of these variations represent a process different than the one causing pure isolated CIDP remains to be clarified. A small number of these patients have a malignant plasma cell dyscrasia and an immunoglobulin G (IgG) (or less commonly IgA) paraprotein that causes the POEMS syndrome. Most patients with CIDP-MGUS and an IgG or IgA monoclonal protein have a disorder that is indistinguishable from idiopathic CIDP, including improvement with immune therapies and preferential involvement of the nerve roots.
9.2.1.3 Treatment
CIDP often responds to treatment, underlining the importance of establishing the diagnosis. Reports of improvement with corticosteroids in 39–95 % of patients (Dyck et al. 1975; Prineas and McLeod 1976; McCombe et al. 1987) have withstood controlled testing (Dyck et al. 1982a). Patients can, however, improve spontaneously. Plasma exchange (Dyck et al. 1986) and intravenous immunoglobulin administration (Faed et al. 1989; Van Doorn et al. 1991) appear to be effective, clinical benefit usually becoming apparent within days to weeks.
9.2.2 Pathology
9.2.2.1 General Considerations
Multifocal peripheral nerve disease that predominantly involves the proximal nerve (root, plexus, large nerves) is the substrate of CIDP (Milder et al. 1985; Dyck et al. 1975; Thomas et al. 1969). Examination of the sural nerve only provides a keyhole view of the entire picture. Nevertheless, since the clear delineation of this treatable neuropathy in the 1970s, biopsy has been used to confirm the diagnosis and justify the treatment required. In recent years in our institution, a frequent indication for sural nerve biopsies was for confirmation of a clinical diagnosis of CIDP. Other workers describe a similar experience (Krendel et al. 1989; Solders 1988; Oh 1990).
An understanding of the information to be gained from nerve biopsy in CIDP leads one to question the value of this procedure in this setting. The rationale for biopsy has not been clearly defined but relates to corticosteroids being the mainstay of therapy and the clinicians’ desire to have proof of the diagnosis before undertaking a potentially complicated long-term treatment (Krendel et al. 1989). However, an entirely normal sural nerve can be seen in as many as 24 % of cases, predominantly axonal pathology is not unusual, and often nerve biopsy changes are nonspecific (Tables 9.1 and 9.2). Inflammation is seen in two-thirds or less of the specimens, and a characteristic inflammatory predominantly demyelinating picture is probably present about half of the cases (Table 9.1). We have examined some biopsies in which only chronic axonal degeneration was seen despite an extensive search for inflammation or demyelination, where the patient met clinical criteria for CIDP and responded to immune-modulating treatment.
Table 9.1
Biopsy results in literature CIDP series
Biopsy shows:
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|
