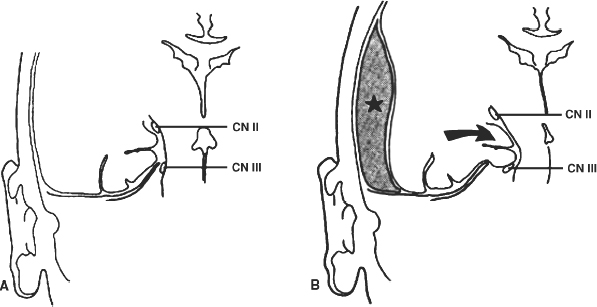
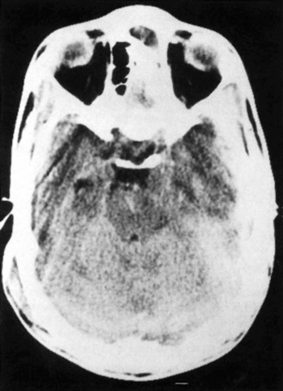
4 The Recognition and Management of Cerebral Herniation Syndromes Brian T. Andrews One of the most abrupt and dreaded complications of intracranial pathology arises when there is herniation of brain tissue across the natural boundaries of dura and bone. This usually occurs when an expanding mass lesion exhausts the capability of the brain and cerebrospinal fluid (CSF) to tolerate added volume, resulting in elevated intracranial pressure (ICP). Herniation may also occur due to local mass effect, such as in the temporal lobe or posterior fossa, without a major rise in overall ICP, or when differential CSF pressures exist across anatomic barriers, such as low intraspinal fluid pressure. Herniation generally leads to anatomically characteristic syndromes, including transtentorial and cerebellotonsillar herniation, although the clinical manifestations depend on the speed at which herniation occurs. A slowly expanding mass lesion such as a chronic subdural hematoma or gradually enlarging tumor may lead to severe anatomic herniation, with few initial neurological findings and little direct morbidity.1,2 In contrast, a rapidly expanding mass lesion or shifting pressure gradient usually results in a profound and often devastating progression of neurological deficits, with a high risk of morbidity and mortality, if not quickly recognized and effectively treated.3,4 This chapter will focus on the recognition and management of these rapidly evolving cerebral herniation syndromes. The most common causes of herniation are intracranial hemorrhage of traumatic or spontaneous origin1,2,3,5,6 Regional or diffuse brain edema caused by cerebral ischemia and infarction are also common.2,6,7 In each case the herniation syndrome may involve structures above the tentorium, in the posterior fossa or in both spaces. In addition there are a wide number of other causes for cerebral herniation, such as acute hydrocephalus,7 hepatic encephalopathy,8 tumor enlargement with associated vasogenic edema,9 and therapeutic lumbar CSF drainage.4 The most common anatomic sites of herniation are across the tentorium cerebelli, either downward or upward,6 and downward across the foramen magnum.4 In all of these situations the neurosurgeon must be adept at the recognition and management of these important complications of central nervous system pathology. The tentorium cerebelli is an arched lamina of dura that lies in the cerebrocerebellar fissure. Elevated in the midline and sloping downward to attach to the petrous bone laterally and the transverse grooves of the occipital bone posteriorly, the slightly concave surface of concentric, circumferential, and radial dural bands yields little to pressure. It has been described as a “mechanically perfect means of directing forces away from the vulnerable midbrain,”10 which passes through the incisura of the tentorium. The incisura, or tentorial notch, extends from the edges of the tuberculum sella back to the confluence of the straight sinus and the great vein of Galen. The space between the free edge of the tentorium and the lateral border of the midbrain, forming the ambient cistern, varies in size from virtually no space, with direct contact of the midbrain and dura in up to 43% of postmortem specimens, to as much as 7 mm of space on either side.11 The medial margin of the uncus of the temporal lobe usually overhangs the edges of the incisura, and closely approximates the more medial structures. Adler and Milhorat12 have classified the dimensions of the tentorial notch into eight types, and noted that the amount of exposed cerebellar parenchyma within the notch and the relationship between the brainstem and the tentorial edge and brainstem position varied greatly among individuals, potentially altering susceptibility to transtentorial herniation from a supratentorial or infratentorial source. Critical structures within the incisura include the third (oculomotor) cranial nerves, the posterior communicating and posterior cerebral arteries (PCAs), and the midbrain. The third cranial nerves emerge from the medial aspect of the cerebral peduncles to pass through the subarachnoid space over the posterior clinoid processes anterolaterally to enter the dura at the superior margin of the cavernous sinuses. The medial margin of the uncus is immediately lateral to the third nerve in its subarachnoid course. Third nerve length, trajectory, and anatomical relationship to the skull base varies widely among individuals.13 The pupillo-constrictor fibers run along the periphery of the third nerve, and are exquisitely sensitive to external pressure.12 Thus mass effect, which compresses the uncus against the nerve, or pressure from below which stretches or kinks it against the dural edge, results in loss of constriction, the resulting pupillary dilation being the hallmark clinical sign of transtentorial herniation.2,6,10–13 Superior and lateral to the third nerves are the paired posterior communicating arteries, arising anteriorly from the internal carotid arteries to run back to join the PCAs, which arise from the distal bifurcation of the basilar artery (BA). The paired PCAs course laterally over the oculomotor nerves and the free edge of the tentorium, rendering them extremely vulnerable to occlusion by downward pressure. Inferiorly, the paired superior cerebellar arteries arise from the BA to course laterally under the tentorium; these are vulnerable to occlusion from upward herniation from the posterior fossa. Within the incisura is located the midbrain, consisting of the cerebral peduncles anteriorly, the midportion or tegmentum, and posteriorly the tectum, comprised of the superior and inferior colliculi. Through this region pass all of the fiber tracts that connect the cerebral cortex, basal ganglia, thalamus, and upper brainstem nuclei with the lower brainstem and spinal cord. Also within this region are the oculomotor and trochlear nerve nuclei, the substantia nigra, the red nuclei, the periaqueductal gray matter, and the neurons of the reticular activating system (RAS). The proximal aqueduct of Sylvius passes centrally here from the posterior third ventricle, rendering a high risk of obstructive hydrocephalus from mass effect in this area. The blood supply to the midbrain consists of interpeduncular arteries from the distal BA and the proximal PCAs; these give rise to smaller, perforating arteries. Inferiorly, small circumferential arteries arise from the BA to irrigate the outer substance of the midbrain. These perforating arteries are all functional “end-arteries” with few collaterals within the midbrain parenchyma. This becomes important when mechanical compression causes occlusion of these small vessels, leading to severe local ischemia. The subarachnoid spaces of the incisura are divided into several cisterns, which may initially act as hydraulic buffers protecting the midbrain.10,13,14 The interpeduncular cistern lies anteromedially to the cerebral peduncles, just above the prepontine cistern in the posterior fossa; others have described these together as the “basal cistern.”10 Lateral to the midbrain lies the ambient or perimesencephalic cistern. Radiographic evidence of compression or effacement of the ambient cistern gives verification of transtentorial herniation.1,14 Compression of this cistern on the initial computed tomography (CT) of the brain has been shown to have a negative impact on prognosis in the setting of intracerebral hematoma15 and of head injury16; in both cases preservation of the cistern correlates to a far better likelihood of good outcome than for those patients with compression of one or both cisterns. Posterior to the midbrain is the quadrigeminal plate cistern, also known as the “cistern of the vein of Galen.” First described anatomically in 1896,17 transtentorial herniation is the medial and caudal dislocation of brain parenchyma from the supratentorial space through the incisura (Fig. 4-1). Classic pathological studies in the 1920s by Meyer18 document medial displacement of the uncus, obliteration of the ambient cistern, compression and displacement of the oculomotor nerve, and midbrain. A deep groove is often formed along the undersurface of the ipsilateral uncus by the firm edge of the tentorium. Experimental models have since demonstrated that expansion of an intracranial mass above the tentorium results in a gradient of increased ICP, highest ipsilateral to the mass and above the tentorium, less below the tentorium, and lowest in the spinal subarachnoid space.19 The result is medial herniation of the uncus of the temporal lobe into the ipsilateral ambient cistern, with stretching, torsion, and compression of the oculomotor nerve, compression of the midbrain itself, and occlusion of the aquaduct of Sylvius.10,20–22 The stalk of the pituitary gland may be stretched across the diaphragma sellae, causing infarction of the pituitary gland itself.21 Computed tomography shows in severe herniation that the midbrain is rotated or twisted, and the cerebral peduncles elongated and flattened14 (Fig. 4-2). Figure 4-1 Coronal view of the temporal lobe and adjacent midbrain. (A) The normal relationships, with preserved ambient cistern. (B) The appearance of transtentorial herniation, with downward displacement of the brain stem, medial displacement of the uncus, and compression of the oculomotor nerve and midbrain structures. Figure 4-2 Axial CT in a patient with severe transtentorial herniation shows the midbrain to be compressed and rotated by the medial deviation of the left uncus Due to contralateral displacement of the brainstem and compression of the opposite cerebral peduncle against the tentorium, Kernohan and Woltman22 in 1929 identified a notching of the opposite side of the midbrain, later to be identified both clinically and pathologically as the “Kernohan’s notch phenomenon.” There may also be downward displacement of the brainstem. Using magnetic resonance imaging, Reich et al1 showed that fully one half of patients with transtentorial herniation also had downward shift and concurrent cerebellar tonsillar herniation at the foramen magnum. Ropper23 has shown however, that the clinical syndrome can evolve with only horizontal displacement of the brainstem and little or no downward displacement. Reich et al1 also showed, using MRI, that in more chronic cases, radiographic evidence of herniation could precede clinical manifestations, and identify patients with earlier, reversible clinical findings, and that resolution of clinical herniation was accompanied by reversal of radiographic findings. Transtentorial herniation causes distortion of the arteries of the posterior circulation, with stretching and occlusion of the small perforating branches that supply the upper brainstem.10,21 This may result in rupture of these small arteries, with consequent brainstem hemorrhage (Duret’s hemorrhages). Hemorrhages may also occur because of initial ischemia due to vessel occlusion from downward displacement, followed by reperfusion of the infarcted areas as the displaced tissue relaxes. The PCAs are commonly occluded as they cross the incisura, resulting in characteristic infarction of one or both occipital lobes as an added complication. Histologic changes include lipid vacuolization within the herniated uncus, with neuronal swelling and peripherally displaced nuclei. With time, surviving neurons become pyknotic and a fibrous gliosis may develop in survivors of the clinical syndrome. Edema also occurs within the brainstem, accompanied by the neuronal and white matter changes of ischemia. Thrombosed veins, venulae, and capillaries are seen, attributed to both direct compression and ischemia.10 The classic signs of transtentorial herniation include the triad of anisocoria, with initial ipsilateral pupillary dilation, often irregular in shape,24 loss of the light reflex, alteration in level of consciousness, and an asymmetric motor response, usually a contralateral hemiparesis. As herniation progresses, the pupillary dilation becomes bilateral and the pupils fixed and nonreactive to light.2,6,10–12,18,22 Alteration in level of consciousness is usually progressive to the point of coma, from the effect of the mass lesion on ICP, global dysfunction of the cerebral hemispheres, and compression of the RAS of the midbrain. The hemiparesis is usually contralateral to the side of the mass lesion due to compression of the ipsilateral cerebral peduncle, and may initially be mild, but usually worsens to a hemiplegia as brainstem compression progresses. In ~25% of cases the hemiparesis is ipsilateral to the dilated pupil, due to midbrain shift and compression of the contralateral cerebral peduncle against the opposite tentorial edge, termed the “Kernohan’s notch phenomenon.”22 Pupillary size and reactivity depend upon a balance between the effects of the sympathetic and parasympathetic nervous systems on the pupils. Sympathetic innervation arises from the hypothalamus and brainstem, passing through the cervical spinal cord to synapse in the intermediolateral tract of the upper thoracic spinal segments. Preganglionic fibers pass through the ventral roots of the upper thoracic spinal cord to ascend through the inferior and middle cervical sympathetic ganglia to synapse in the superior cervical ganglion. Postganglionic fibers then ascend along the internal carotid artery to enter the orbit through the superior orbital fissure with the nasociliary nerve. Fibers then enter the globe as the long ciliary nerve. Sympathetic discharges not only innervate the dilator pupillae muscles but also innervate the smooth muscle of the levators of the eyelid (Muller’s muscle). Parasympathetic innervation arises from the Edinger-Westphal nucleus, dorsal to the oculomotor nucleus in the midbrain. The preganglionic fibers travel on the periphery of the oculomotor nerve as it passes forward from the interpeduncular fossa to enter the dural edge of the incisura and the cavernous sinus. These fibers are exquisitely sensitive to mechanical stretch or compression. After the oculomotor nerve enters the superior orbital fissure, the parasympathetic fibers pass to the ciliary ganglion and synapse. The postganglionic fibers form the short ciliary nerve which enters the sclera to innervate smooth muscle fibers that constrict the pupil. Transtentorial herniation of the uncus results in both direct compression and stretching or torsion of the ipsilateral oculomotor nerve itself and then compression of the oculomotor and Edinger-Westphal nuclei in the midbrain. These result in progressive loss of parasympathetic tone, with continued sympathetic innervation resulting in an enlarging and often initially irregular ipsilateral pupil.10,15,19,24 Marshall et al24 have shown that relatively mild increases in ICP alone may result in an irregular or dilated ipsilateral pupil. As midbrain compression and ischemia progresses there may be loss of both parasympathetic and sympathetic innervation bilaterally, resulting in midposition (4 to 5 mm) pupils that are fixed to light. Marshman et al25 have shown that the dilated and fixed pupil can rarely be contralateral to the mass lesion and thus “false-localizing,” possibly due to stretching of the contralateral oculomotor nerve from hemispheric mass effect and midline shift of structures well above the midbrain. With increased pressure on the oculomotor nerve and nucleus, loss of ipsilateral extraocular movements may occur, with resulting tonic deviation of that eye laterally due to continued abducent nerve function. Other ocular findings may also be noted, such as ptosis and impaired vertical or upward gaze due to compression of the dorsal midbrain.1, 10,24 In humans level of consciousness reflects both level of arousal or alertness, and the presence of conscious behavior or cognitive function. Normal arousal relies upon intact function of the RAS, whereas conscious behavior reflects function of the cortical hemispheres. The RAS is a diffuse network of neurons that form a central core of the brainstem, most prominent in the midbrain. The RAS is not distinct, and its neurons are extensively interconnected, with collateral input from every major sensory pathway, particularly the spinothalamic tract and the trigeminal nerve. Numerous connections ascend into the subthalamus, thalamus, hypothalamus, and the basal forebrain structures including the limbic system. Other connections extend diffusely and reciprocally into the neocortex. Stimulation of the RAS produces a general activation of the cerebral cortex, in part by abolishing inhibitory input from the thalamus and the limbic system. Arousal or alertness is dependent upon the integrity of the RAS. Thus, with either direct compression or ischemia of the midbrain there is a loss of RAS function and decrease in alertness and level of consciousness. Usually the lesion that has resulted in herniation has also affected the cortical hemispheres, either directly or through global elevation in ICP, resulting in a decrease in conscious behavior and cognitive function. Cortical lesions of increasing size usually result in a progressive decrease in level of alertness and cognitive function, associated in part with the degree of midline brain shift.1,2,15 Alteration in the level of consciousness, a hallmark sign of transtentorial herniation, thus may result from either compression of the midbrain, affecting the function of the RAS, or dysfunction of the cerebral hemispheres in a localized or diffuse fashion. Asymmetric motor findings are the third clinical manifestation of transtentorial herniation. Most often hemiparesis is due to compression of the corticospinal tracts of the ipsilateral cerebral peduncle, and thus is contralateral. However, motor paresis may also result from direct compression of the ipsilateral hemisphere itself. As noted above, in ~25% of cases the hemiparesis is ipsilateral to the side of herniation and the dilated pupil, due to midbrain shift to the opposite side and compression of the contralateral cerebral peduncle against the opposite tentorial edge, the Kernohan’s notch phenomenon.23 A similar clinical condition occurs when a mass lesion within the posterior fossa results in herniation of brain tissue upward through the incisura, resulting in acute impaction and compression of midbrain structures 1,6,11,15. The usual causes are hematomas and infarctions and other mass lesions within the cerebellum such as abcess and tumors,1,6,11,15 along with more unusual causes such as adipose graft prolapse into the posterior fossa following translabyrinthine craniotomy.26 The usual mechanism is elevated pressure within the narrow confines of the posterior fossa, with upward displacement of the cerebellar vermis, compressing the dorsal midbrain within the incisura.1,6,11 Upward herniation is more likely when the mass arises within the vermis itself, or when the incisura is large in size.6 There may also be exacerbation of the pressure gradient across the tentorium by placement of a shunt or ventriculostomy above for control of secondary hydrocephalus and ICP monitoring.1,6 Pathologically there is compression and distortion of the midbrain, compression of the aqueduct of Sylvius, buckling of the quadrigeminal plate, and displacement and occlusion of the vein of Galen. This venous obstruction may cause secondary hemorrhagic infarction of the diencephalons.1,6 The distal branches of the superior cerebellar artery may become compressed against the underside of the tentorium resulting in ischemia, edema, and infarction of the cerebellar hemispheres, aggravating the condition 1,6. The clinical manifestations of upward transtentorial herniation vary, and in some ways are dissimilar to those of downward herniation. Level of consciousness may deteriorate to coma, often associated with small, minimally reactive pupils (so-called “pontine-pupils”).6,11,15 These pupillary changes are due to direct compression of the pons, with parasympathetic papillary input from the midbrain unopposed by sympathetic tone descending through the pontomedullary region.11,12 Cuneo et al,6 however, have also described the initial development of anisocoria and fixed, midposition, or even large pupils due to evolving distortion and compression of the midbrain and oculomotor nerves themselves. Also characteristic of upward herniation is the absence of vertical eye movements due to pretectal compression. There may also be conjugate downward deviation of the eyes, or skew gaze.1,11 Flexor or extensor posturing on motor response1,6,15 and Cheyne-Stokes respirations or hyperventilation can be noted as well. Reich et al1 have identified upward herniation on MR imaging in the sagittal plane as a cephalad deviation of the proximal opening of the aqueduct of Sylvius, above the level of the incisural line. They have also shown angulation or buckling of the quadrigeminal plate and ventral bowing and displacement of the brainstem. They noted that MR imaging may identify upward herniation prior to the development of extreme neurological consequences, and that such imaging can be used to follow the course of progression or recovery of upward herniation, which correlates closely with clinical progression or recovery. Herniation of the cerebellar tonsils downward through the foramen magnum is the final type of brain herniation that may have immediate and devastating neurological consequences.1,6,10,15,18,22 Most often occurring as a result of a mass in the inferior cerebellum, the cerebellar tonsils are displaced downward through the foramen magnum, resulting in direct compression, ischemia, and infarction of the tonsils themselves, compression of the medulla oblongata, and obstruction of the foramina of Luschke and Magendie.6 Another result of cerebellotonsillar herniation is direct compression of the pons and medulla against the clivus, with distortion or closure of the fourth ventricle. Closure of any of the ventricular outflow tracts may lead to obstructive hydrocephalus, which can further increase ICP, both above and below the tentorium.6 Cerebellotonsillar herniation may also occur due to a large supratentorial mass lesion, which causes elevated ICP and downward displacment of the entire brainstem 18. After lumbar puncture (LP), with a lowered CSF pressure below the foramen magnum and an enhanced rostral-caudal pressure gradient, a supratentorial mass may result in cerebellotonsillar herniation and an abrupt clinical decline. Jennet and Stern18 showed experimentally that a large supratentorial mass lesion was associated with mechanical distortion and downward displacement of the brainstem, and tonsillar impaction into the foramen magnum. Reich et al1 have shown, with sagittal MR imaging, that anatomic herniation at the foramen magnum often accompanies transtentorial herniation due to a supratentorial mass, and that such herniation may be reversed as the mass is treated and clinical herniation resolves. The pathologic consequences of cerebellotonsillar herniation include direct mechanical compression of the medulla oblongata against the lower clivus and anterior foramen magnum, often resulting in a transverse groove along the ventral medulla.1,6 Ischemia and infarction of the cerebellar tonsils, lower cerebellum, and the entire lower brainstem and upper spinal cord may occur due to occlusion of the vertebral arteries, their branches, and the origin of the anterior spinal artery.1,6 Histologic changes include edema and lipid vacuolization within the herniated and compressed tissues, along with pyknotic nuclei and poorly staining cytoplasm within the neurons of the brainstem nuclei. Clinically, rapid descent of the cerebellar tonsils and impaction of the medulla oblongata may cause sudden apnea and circulatory collapse.1,6 Subsequent coma is more often due to respiratory and circulatory arrest than to the brainstem compression itself. Clinical signs that may precede such collapse include those of pontomedullary compression, including pontine pupils, loss of lateral eye movements, or internuclear ophthalmoplegia due to dysfunction of the abducent nerve nuclei and parapontine reticular formation. Some preservation of vertical eye movements may be retained as upper brainstem function remains intact, and “ocular bobbing” may be noted.6 The motor signs of pontomedullary compression may include extensor posturing, but more immediate flaccid quadriplegia occurs due to compression of the descending medullary corticospinal tracts. Respiratory changes may include immediate apnea, cluster breathing, gasping, and ataxic breathing patterns, but not the more familiar Cheyne-Stokes respirations, which are characteristic of hemispheric or midbrain-diencephalic insults. Given the often rapid onset of abrupt and profound cardiopulmonary collapse with cerebellotonsillar herniation, it is critical that any neurological deterioration that includes potential signs of such herniation be immediately recognized and action taken to stabilize the patient and lower ICP. This should be followed immediately by diagnostic measures to allow definitive treatment of the intracranial mass lesion causing the decline. The possible hazards of LP in a patient with clinical papilledema were recognized soon after the introduction of this procedure into clinical practice.27,28 The occurrence of transtentorial herniation, or more commonly, cerebellotonsillar herniation, in a patient with an intracranial mass lesion may occur within minutes of removing CSF from the lumbar region, or may be delayed for hours or longer. The mechanism of herniation results from an increase in both brain volume and ICP. With a decrease in CSF pressure below the foramen magnum there is rostralcaudal displacement of brain tissue. Herniation occurs only in the presence of some degree of obstruction of normal CSF flow between the cranial and the spinal subarachnoid space.27 When there is normal free flow, the fall in lumbar CSF pressure can equilibrate with the intracranial cavity without brain displacement. Lumbar puncture may also exacerbate an already existing or impending herniation syndrome that is causing blockage of the subarachnoid space, either at the incisura or the foramen magnum, with clinical signs of herniation occurring only after the puncture has been performed. Cerebral herniation after an LP is, in fact, a rare event. A large early clinical series and review of earlier reports have indicated that the incidence in patients with elevated ICP was less than 1.2%.28 More recently Duffy29 reported that 7 of 52 patients with acute subarachnoid hemorrhage deteriorated clinically at the time that an LP was performed; this may have had more to do with aneurysm rebleeding in this series than with herniation occurring as a result of the puncture. The risks of cerebral herniation make it imperative that diagnostic imaging such as CT scan be performed prior to an LP in any patient with a suspected intracranial mass or elevation of ICP.27 In the presence of a significant mass above or below the tentorium, midline shift, or noncommunicating hydrocephalus, an LP should be avoided. If there is little mass effect and the results of CSF analysis are important to the clinical diagnosis, then an LP should be performed, understanding the possibility of what is, in fact, a very unlikely complication. An acute herniation syndrome at the foramen magnum has recently been described due to the use of perioperative lumbar drainage in three patients.4 The authors describe the development of an “acquired Chiari malformation” and the development of a negative pressure gradient between the cranial and the spinal subarachnoid spaces. Because the definition of cerebral herniation relies upon the bedside neurological examination, it is imperative that these clinical findings accurately reflect intracranial pathology. Severe systemic hypotension, hypoxia, or hypothermia can all depress neurological function and confound the diagnosis of cerebral herniation syndromes. Systemic hypotension is a common complication of severe head injury which can markedly worsen outcome and decrease chances of survival.30–33 Andrews et al31 reviewed a series of 36 patients to analyze the effects of severe hypotension or preceding cardiac arrest at the time of the initial neurological examination following head injury. Each patient had neurological signs at the time of admission that might be consistent with a herniation syndrome. Ten patients had been successfully resuscitated from cardiac arrest, 7 had an initial systolic blood pressure (SBP) of less than 60 mm hg, and 19 had an initial blood pressure of 60 to 90 mm Hg. The median Glasgow Coma Score was 3 (range, 3 to 8), and the neurological findings for each group were similar. Among the 10 patients with resuscitated cardiac arrest, 4 (40%) had anisocoria and 6 (60%) had bilaterally fixed and dilated pupils; all 10 had absent corneal reflexes. Nine patients (90%) were flaccid and one (10%) had bilaterally extensor posturing. Of the 7 patients with an initial SBP of less than 60 mm Hg, 2 (29%) had anisocoria, all had absent corneal reflexes, and all were flaccid. Of the 19 patients with an initial SBP of 60 to 90, 9 (47%) had anisocoria, 8 had active corneal reflexes; 4 had hemiparesis, 4 exhibited extensor posturing, and 11 (58%) were flaccid. Each patient underwent surgical exploration and/or radiographic assessment for an underlying structural lesion causing the apparent herniation syndrome. Only 1 (10%) of the 10 patients resuscitated from cardiac arrest had a significant mass lesion; only 1 (14%) of the 7 with initially severe hypotension had a hematoma. In neither group were the findings of the initial clinical examination useful in identifying the presence or site of an intracranial mass lesion. In contrast, of the 19 patients with an initial SBP of 60 to 90 mm Hg, 13 (68%) had extra-axial hematomas (p <0.01), including 78% of those with initial anisocoria. In each case the hematoma was ipsilateral to the dilated pupil (p <0.05). This study indicates that an initial SBP of at least 60 mm Hg is needed to perfuse the brain adequately and allow the neurological examination accurately to reflect intracranial pathology. Among patients with more profound hypotension, or initial cardiac arrest, the findings of the neurological examination reflect diffuse cerebral ischemia, not herniation. Systemic hypoxia is an even more common complication of severe head injury than hypotension,30,31,33 occurring in 30% or more of patients at the time of initial evaluation. The effect of hypoxia on the neurological examination is often complicated by systemic hypotension, which occurs due to hypoxic effects on the myocardium and peripheral vasculature. If hypotension is prevented, normal humans can tolerate an extremely low PaO2 without major neurological manifestations or sequelae. Gray and Horner34 reported that among 22 patients with a PaO2 of 20 mm Hg or less, 8 remained alert, 7 somnolent, and 7 comatose. Severe hypoxia usually causes clinical signs of a metabolic encephalopathy, with deterioration in level of consciousness to eventual coma, along with changes in respiratory pattern, tremor, asterixis, myoclonus, and flexor or extensor posturing.34,35 Brainstem reflexes usually remain intact until profound anoxia has occurred, at which time there is papillary dilation and loss of the oculocephalic reflexes.35 It is important to recognize that any of a wide variety of metabolic insults like hypoxia can further depress the neurological examination, particularly in the severely injured brain. Such problems as hypothermia, severe hyper- or hypoglycemia, hyponatremia, or drug intoxications may alter level of consciousness,35 and should be considered when first evaluating any patient in coma with or without evidence of brainstem dysfunction, especially when the clinical history is unclear. Treatment of acute cerebal herniation must begin upon recognition of the clinical condition and be concurrent with diagnostic studies being completed. Prolonged or persistent herniation will lead to irreversible ischemic damage to the deep midline structures of the cerebral hemispheres and the brainstem, resulting in permanent morbidity or death. The immediate goals include the reduction of elevated ICP while maintaining cerebral perfusion pressure (CPP), and oxygenation and prevention or correction of hypercarbia and acidosis.8,36 If the cause of the herniation syndrome is unknown, then a CT scan of the brain performed as an emergency can identify of there is a directly treatable mass lesion. Lowering elevated ICP and supporting blood pressure and oxygenation are essential first steps. Blood pressure management, controlled ventilation, and intravenous infusion of mannitol are the primary means of achieving these goals. These measures allow the brain temporarily to accommodate an underlying cause of increased ICP such as a mass lesion, until definitive diagnosis and treatment can be initiated. The initial steps required to adequately resuscitate a patient with acute cerebral herniation are the same regardless of the cause, be it severe head injury, intracranial hemorrhage or diffuse cerebral edema. Key to resuscitation are the ABCs: airway, breathing, and circulation. First, the patient must have an adequately protected and controlled airway. In the field, initial mask ventilation with 100% oxygen usually suffices, although often now trained prehospital staff may successfully provide orotracheal intubation before the patient arrives. Once the patient is in the emergency department, prompt endotracheal intubation should be provided if it has not already been performed. In patients with head injury, a lateral cervical radiograph should be obtained first to rule out an obvious cervical fracture or instability. Even with a negative radiograph, only gentle axial traction should be provided during intubation and extreme extension or distraction of the cervical spine should be avoided, as there is a 20% chance of significant injury despite a normal screening lateral radiograph.37 Alternatively, nasotracheal intubation (if a major base of skull fracture is not suspected), or cricothyroidotomy may be performed. Intubation also remains a critical first step in the already hospitalized patient who develops signs of cerebral herniation, such as after mild or moderate closed head injury or after cranial surgery. Once an airway has been established, controlled ventilation with 100% oxygen should be maintained with the goals of improving arterial oxygenation and reversing hypercarbia and respiratory acidosis 36. Hyperventilation can provide an immediate decrease in arterial carbon dioxide tension (PaCO2), which increases the pH of blood and causes a respiratory alkylosis. This results in diffuse cerebral vasoconstriction, decreasing cerebral blood volume and lowering ICP. In patients with expanding hematomas causing transtentorial herniation, hyperventilation can temporarily result in a reversal of pupillary anisocoria as well as hemiparesis, while diagnostic studies can be performed and the hematoma identified and treated.36,38 This is the only setting in which the Brain Trauma Foundation, American Association of Neurological Surgeons (AANS) Joint Section of Neurotrauma, and Critical Care Guidelines for the Management of Severe Head Injury support the use of hyperventilation.4 The risk of hyperventilation is the induction of cerebral ischemia due to excessive vasoconstriction,36 and thus as soon as diagnostic studies have been performed, if a mass lesion is identified, this should be immediately addressed and PaCO2 normalized. If diagnostic studies do not identify a mass lesion, then one should bring the PaCO2 to 30 to 35 mm Hg, unless further hyperventilation is guided by the use of additional monitoring to avoid ischemia, such as AVDo2 measurement.39 The vasoconstriction induced by hyperventilation is effective only in regions of the brain where cerebrovascular CO2 responsiveness remains intact; therefore ICP may respond less to hyperventilation in patients with diffuse brain injury than in those with more focal abnormalities such as hematomas, where large regions of the brain can still respond.36 Since the latter circumstance is the case for many patients with herniation syndromes, initial hyper-ventilation remains an optimal initial treatment. In patients with diffuse lesions requiring ongoing treatment of elevated ICP, the use of ongoing hyperventilation is much more controversial. Due to the risks of ischemia, and the fact that the vasoconstriction cause by hyperventilation is lost over time40, most practitioners now suggest normalizing the PaCO2 to 30 to 35 mm Hg. Cruz et al,39 however, advocate assessing global cerebral ischemia using AVDo2 monitoring to guide the ongoing use of hyperventilation to the degree that it lowers ICP but does not induce ischemia. The final step of the initial ABC’s is to assess and support the circulation and blood pressure. It is critical that systemic hypotension be prevented or rapidly corrected to maintain brain perfusion. From the start adequate intravenous access must be established. Intravascular volume resuscitation should be provided as needed to stabilize or maintain blood pressure, using a balanced salt solution such as Ringer’s lactate solution. If the blood pressure is initially normal, hydration should be moderated to avoid overhydration, which may aggravate cerebral edema or lead to pulmonary edema. In the head-injured patient, the most common cause of systemic hypotension is hemorrhagic shock. In this case volume resuscitation should include the use of blood products.41 Patients with multisystem injuries may have additional reasons for hypotension, such as low cardiac output due to a cardiac contusion or tamponade.41 They may also show loss of systemic vascular resistance due to a spinal cord injury. These alternatives should be considered if the blood pressure does not respond to initial volume resuscitation, or if the clinical picture does not fit with that of hemorrhagic shock.41 As hemorrhagic shock is being treated with volume expansion, using crystalloids and blood products, the source of blood loss must be quickly found and controlled. Common sites of hemorrhage include the chest and abdomen, pelvis, and long bone fractures.41 These problems must be addressed by the appropriate surgical specialist concurrent with management of the severe head injury. There has been some enthusiasm for the use of hypertonic saline to initially resuscitate patients with severe head injury, and to treat elevated ICP.42 It was demonstrated that hypertonic saline solutions are effective in reducing ICP and usually improve CPP, but there was no notable superiority of such solutions to the use of conventional resuscitation and infusions of intravenous mannitol.43 Qureshi et al44 described the successful reversal of herniation due to supratentorial mass lesions using combinations of hypertonic saline and mannitol, barbiturates, and hyperventilation, but no clear role of hypertonic fluids in the initial resuscitation of patients with herniation has been established. In patients with hemorrhagic shock and herniation, the use of hypertonic saline may be more appropriate, given the contraindication of use of mannitol in such patients.42,43 Additional steps to resuscitate a patient with acute cerebral herniation include the use of intravenous mannitol. Except in the setting of hemorrhagic shock, the author recommends the immediate bolus infusion of mannitol, 1.0 to 1.5 gm/kg body weight.5,36 Mannitol, a six-carbon sugar similar to glucose, is not metabolized, nor does it cross the blood-brain barrier. It remains predominantly in the intravascular space, and causes a direct vasoconstriction due to its effects on blood viscosity.45–47 It also has effects on red blood cell deformability and hemodilution and improved red blood cell oxygen transport. 46 All of these result in a near-immediate decrease in brain volume, improvement in intracranial compliance, and lowered ICP.48 Mannitol improves bloodflow to all parts of the brain, including the brainstem.40 Finally, mannitol results in a somewhat more delayed osmotic dehydration of the brain. Because of the cardiovascular effects of mannitol infusion, its use is generally contraindicated in the setting of cardiovascular instability or hemorrhagic shock.41 To avoid the risk of hypotension from rapid infusion of mannitol, it should be given at a rate no higher than 0.1 gm/kg/minute.49 The dosage of mannitol infusion in the initial management of severe head injury has historically ranged between 0.25 and 1.5 grams/kg body weight.3,36 The author has generally used dosages in the higher range for the initial bolus infusion. Recently Cruz et al,3 in a prospective randomized Class I study, compared initial use of mannitol, using “conventional dosage” and “high” dosage in patients with documented subdural hematomas. Patients in the conventional dosage group received 0.6 to 0.7 gm/kg mannitol, and those in the high dosage group received a total of 1.2 to 1.4 gm/kg if they did not have pupillary anisocoria, and 2.2 to 2.4 gm/kg if they had anisocoria. The low-dosage group had significantly worse cerebral oxygen extraction and cerebral swelling than the high-dosage group. Preoperative improvement in anisocoria was also significantly better in the high-dosage group. After six months, the Glasgow Outcome Scale scores were significantly better in the high-dosage group than in those receiving the conventional dosage. The same authors have also shown that high dosage (1.4 gm/kg) of mannitol was more effective than conventional dosages (0.7 gm/kg) for patients with traumatic parenchymal hematomas of the temporal lobe causing abnormal pupillary response.49 These results would seem to support strongly the use of high dosage mannitol in patients with clinical transtentorial herniation, particularly if a hemorrhagic mass lesion has been documented to be present. After the initial management steps outlined above, it becomes imperative that the cause of herniation be identified as quickly as possible and treated directly, if possible. Upon completing endotracheal intubation, controlled ventilation with selective hyperventilation and an infusion of intravenous high dosage mannitol is initiated, and a diagnostic computed tomogram of the brain should be performed immediately in hemodynamically stable patients.36 Computed tomography is extremely sensitive to the presence of acute intracranial hemorrhages and other mass lesions that may cause a herniation syndrome, such as cerebral edema, tumor, or hydrocephalus. Computed tomography should also be performed prior to considering LP in any patient with a suspected mass lesion; the identification of such a mass may make LP contraindicated, unless absolutely necessary. Of patients that are hemodynamically unstable due to traumatic injuries to the chest or abdomen, some must go directly to the operating room for treatment of these life-threatening lesions.41 It may be impossible in such cases to obtain a preoperative CT scan. If the patient has not been resuscitated from initial cardiac arrest, or has not had profound hypotension, in which case the clinical findings of herniation are often false localizing,31 it may be reasonable to consider performing exploratory burr-holes on the side of the dilated pupil.5 Because most traumatic lesions causing herniation are located in the epidural or subdural space, burr-holes placed in the temporal, frontal, and parietal areas will be accurately identified by this rapid technique. In the case of nonlateralizing signs of herniation, the burr-holes should be placed bilaterally. Intraoperative ultrasonic imaging of the brain parenchyma can further enhance the diagnostic yield of exploratory burr-holes, allowing identification of parenchymal hematomas or other mass lesions.50 In patients with initial cardiac arrest or profound systemic hypotension, given the lower incidence of intracranial mass lesions,31 it is not appropriate to perform exploratory burr-holes, but the placement of an ICP monitor is reasonable. If the ICP is low, then no further direct treatment of intracranial physiology is appropriate except for general stabilization of the patient. If the ICP in such a case is markedly elevated, then the surgeon may decide that further surgical exploration and ultrasonic examination of the brain are appropriate to identify a treatable mass lesion. After initial CT scanning, or upon exploratory diagnosis, the presence of a posttraumatic mass lesion should lead to immediate surgical evacuation, if feasible.3,5,15,36,49 The presence of residual cerebral swelling after evacuating the hemorrhage should lead to consideration of either an appropriately located lobectomy,51 a large, decompressive craniectomy,52 or both. Decompressive craniectomy has also become a recognized treatment for herniation caused by hemispheric infarction, particularly in the nondominant hemisphere.53 When a decompressive craniectomy is performed, it should be as large as is feasible, to fully decompress the entire hemisphere of the brain, and the bone should generally be stored in a sterile environment at minus 70°C.52 In addition to the above-noted surgical interventions, ICP monitoring should be established at this time for subsequent patient management.36 Immediate surgical management of nontraumatic lesions causing herniation is also often indicated. This may include hemorrhage evacuation for lobar or nondominant hemispheric spontaneous hemorrhage,15 and immediate percutaneous ventricular drainage for hydrocephalus.7 In the setting of upward tentorial herniation or tonsillar herniation at the foramen magnum, emergency management should also include evacuation of responsible mass lesions,6,9 and posterior fossa decompression as needed to decompress the cerebellum and brainstem.4 There may also be situations in which surgical management of a mass lesion is not indicated, such as large, deep-seated hemorrhages within the dominant hemisphere of the brain, those in the brainstem, or when a patient is elderly or has a coagulation disorder. Although the overall prognosis for patients with clinical herniation syndromes is poor, it is by no means hopeless. The prognosis for functional recovery may be quite good, particularly among younger patients who exhibit reversal of the clinical signs of herniation with use of mannitol and hyperventilation, and who have an intracranial mass lesion that can be surgically removed. 3,5,32,49,54 For patients with transtentorial herniation following head trauma, the overall mortality rate is ~70%.5 Among 100 such patients treated surgically, 9% had a good recovery and 9% had a moderately good outcome. Those who recovered were generally younger and had a higher initial GCS than those who died or were severely disabled or vegetative.5 Particularly important is the recognition of patients with an initially high GCS who deteriorate to comatose and then herniate; the cause is often a treatable mass lesion, such as acute epidural hematoma; such patients have the best chance of functional survival with rapid resuscitation and corrective treatment. 5,29 After traumatic herniation, age exerts a profoundly negative impact. Quigley et al54 prospectively examined 380 patients with an initial GCS of 3 to 5 after head injury and evaluated the effects of age, GCS, and pupillary reactivity. They noted that when one or both pupils were nonreactive, all 96 patients older than 50 years of age, and all but one of 121 patients older than 40 years of age, were finally dead or vegetative. Among those under 20 years of age with one or both pupils nonreactive, 11 of 72 (15%) had a functional recovery. For patients with nontraumatic causes of herniation, the prognosis may be much better for functional recovery, as the brain itself may have intact function except for the cause of the herniation syndrome. For patients with acute hydrocephalus,7 tumor-related cerebral edema,9 temporal lobar hemorrhage,15 hemispheric infarction52 or cerebellotonsillar herniation from lumbar drainage,4 appropriate resuscitation and corrective reversal of mass effect can result in a satisfactory outcome. 18. Meyer A. Herniation of the brain. Arch Neurol Psychiatry 1920;4:387–400 20. Jefferson G. The tentorial pressure cone. Arch Neurol Psychiatry 1938;40:857–876 26. Chen TC, Maceri DR, Levy ML, Giannotta SL. Brain stem compression secondary to adipose graft prolapse after translabyrinthyine craniotomy. Case report. Neurosurgery 1994;35:521–524 29. Duffy GP. Lumbar puncture in spontaneous subarachnoid hemorrhage. Br Med J 1982;285:1163–1164 33. Miller JD, Sweet RC, Narayan R, et al. Early insults to the injured brain. JAMA 1978;240:439–442 34. Gray FD Jr, Horner GJ. Survival following extreme hypoxemia. JAMA 1970;211:1815–1817
Anatomy of the Tentorium and Incisura
Biomechanics and Pathology of Transtentorial Herniation
Clinical Signs of Transtentorial Herniation
Pupillary Function
Loss of Consciousness
Hemiparesis
Other Types of Cerebral Herniation
Upward Transtentorial Herniation
Cerebellotonsillar Herniation
Clinical Signs of Cerebellotonsillar Herniation
Cerebral Herniation as a Potential Complication of Lumbar Puncture
The Effect of Hypotension, Hypoxia, and Other Factors on the Neurologic Examination
Cardiac Arrest and Systemic Hypotension
Systemic Hypoxia
Management of Cerebral Herniation Syndromes
Initial Resuscitation and Management
The ABCs
Intravenous Infusion of Mannitol
Subsequent Management
Prognosis in Cerebral Herniation Syndromes
References
< div class='tao-gold-member'>
The Recognition and Management of Cerebral Herniation Syndromes
Only gold members can continue reading. Log In or Register to continue







Full access? Get Clinical Tree


