Chapter 56 Toxic and Metabolic Encephalopathies
Toxic and metabolic encephalopathies are a group of neurological disorders characterized by an altered mental status—that is, a delirium, defined as a disturbance of consciousness characterized by a reduced ability to focus, sustain, or shift attention that cannot be accounted for by preexisting or evolving dementia and that is caused by the direct physiological consequences of a general medical condition (see Chapter 4). Fluctuation of the signs and symptoms of the delirium over relatively short time periods is typical. Although the brain is isolated from the rest of the body by the blood-brain barrier, the nervous system is often affected severely by organ failure that may lead to the buildup of toxic substances normally removed from the body. This is encountered in patients with hepatic and renal failure. Damage to homeostatic mechanisms affecting the internal milieu of the brain, such as the abnormalities of electrolyte and water metabolism associated with renal failure or the syndrome of inappropriate antidiuretic hormone (SIADH) secretion, also affects brain function. In some cases, a deficiency of a critical substrate after the catastrophic failure of an organ, such as hypoglycemia caused by fulminating hepatic failure, is the precipitating factor. Frequently the history and physical examination provide information that defines the affected organ system. In other cases, the cause is evident only after laboratory data are examined.
Clinical Manifestations
Mental status abnormalities are always present and may range from subtle abnormalities, detected by neuropsychological testing, to deep coma. The level and content of consciousness reflect involvement of the reticular activating system and the cerebral cortex. Deficits in the spheres of selective attention and the ability to process information underlie many metabolic encephalopathies and affect performance on many tasks. These deficits are manifested as disorders of orientation, cognition, memory, affect, perception, judgment, and the ability to concentrate on a specific task. Evidence from studies of patients with cirrhosis suggests that metabolic encephalopathies are the result of a multifocal cortical disorder rather than uniform involvement of all brain regions. Abnormalities of psychomotor function may also be present. Among patients with coma of unknown cause, nearly two-thirds ultimately are found to have a metabolic cause. A complete discussion of coma is found in Chapter 5.
Generalized seizures occur in patients with water intoxication, hypoxia, uremia, and hypoglycemia, but only rarely as a manifestation of chronic liver failure. Seizures in patients with liver failure are generally due to alcohol or other drug withdrawal, or cerebral edema associated with acute liver failure. Focal seizures, including epilepsia partialis continua, may be seen in patients with hyperglycemia, and multifocal myoclonic seizures may occur in patients with uremia. Myoclonic status epilepticus may complicate hypoxic brain injury (see Chapter 55).
Toxic Encephalopathies
Hepatic Encephalopathy
A World Gastroenterological Association consensus statement seeks to minimize the substantial confusion in the literature and in clinical practice concerning the diagnosis of HE by using a multiaxial approach (Ferenci et al., 2002). The initial categorization addresses the presence of hepatocellular disease and portacaval shunting. Patients with acute liver disease or fulminating hepatic failure, a disorder occurring in patients with previously normal livers who exhibit neurological signs within 8 weeks of developing liver disease, form the first group. A second group consists of a small number of patients who are free of hepatocellular disease but have portacaval shunting of blood. The largest number of patients have hepatocellular disease with shunts. Further subdivisions address temporal aspects—whether HE is episodic, persistent, or minimal. Causal considerations are then applied to separate patients with precipitated HE from those with recurrent and idiopathic encephalopathy, and to identify the severity of the syndrome. The features that differentiate patients with fulminant hepatic failure from those with the much more common portal systemic encephalopathy are shown in Table 56.1.
Table 56.1 Features Distinguishing Fulminating Hepatic Failure from Chronic Hepatic Encephalopathy or Portal Systemic Encephalopathy
| Feature | Fulminating Hepatic Failure | Portal Systemic Encephalopathy |
|---|---|---|
| HISTORY | ||
| Onset | Usually acute | Varies; may be insidious or subacute |
| Mental state | Mania may evolve to deep coma | Blunted consciousness |
| Precipitating factor | Viral infection or hepatotoxin | Gastrointestinal hemorrhage, exogenous protein, drugs, uremia |
| History of liver disease | No | Usually yes |
| SYMPTOMS | ||
| Nausea, vomiting | Common | Unusual |
| Abdominal pain | Common | Unusual |
| SIGNS | ||
| Liver | Small, soft, tender | Usually large, firm, no pain |
| Nutritional state | Normal | Cachectic |
| Collateral circulation | Absent | May be present |
| Ascites | Absent | May be present |
| LABORATORY TEST | ||
| Transaminases | Very high | Normal or slightly high |
| Coagulopathy | Present | Often present |
Laboratory Evaluations
Neuropsychological tests are an underused and valuable means of diagnosing encephalopathy and monitoring the response to therapy (see Chapter 34). Sixty percent or more of all patients with cirrhosis with no overt evidence of encephalopathy exhibit significant abnormalities when given a battery of neuropsychological tests. Tests of attention, concentration, and visuospatial perception are the most likely to be abnormal. A test battery consisting of Trailmaking Tests A and B, serial dotting, line tracing, and the digit-symbol subtest of the Wechsler Adult Intelligence Scale, Revised, has been recommended for evaluating patients who may have hepatic encephalopathy. This battery is sensitive and relatively specific for the disorder, compared with other metabolic encephalopathies. Patients with alcoholic cirrhosis typically have more difficulty with memory deficits than patients with nonalcoholic cirrhosis. Even though these patients appear to be normal, the degree of impairment, particularly in the visuospatial sphere, may be severe enough to interfere with the safe operation of an automobile or other dangerous equipment. A study comparing patients with minimal encephalopathy with nonencephalopathic patients with cirrhosis and a third group with gastrointestinal disease, found that those with minimal encephalopathy performed the worst during an on-the-road driving test. Specific problems centered on handling, adaptation to road conditions, and accident avoidance. Language functions are usually normal. These data, combined with other studies showing that the quality of life is affected by these abnormalities, suggest that neuropsychological tests should be used more extensively for routine evaluation of all patients with cirrhosis, particularly those without overt evidence of HE.
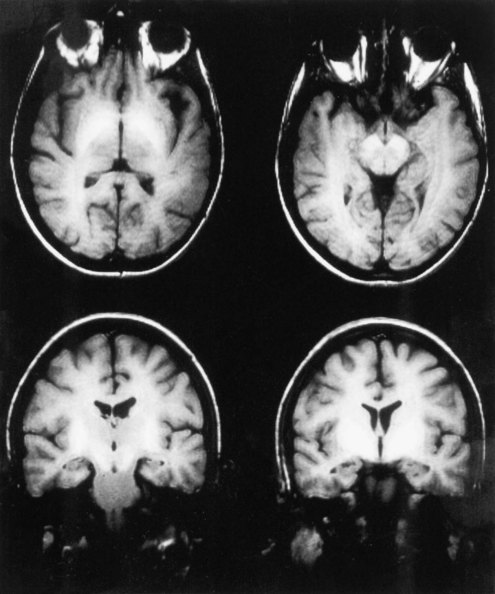
Although the diagnosis of HE is typically made on the basis of clinical criteria, neuroimaging techniques are commonly employed to exclude structural lesions. Magnetic resonance imaging (MRI) and spectroscopic studies have revealed new insights into the pathophysiology of HE (Lockwood et al., 1997). On T1-weighted images, it is common to find abnormally high signals arising in the pallidum. These are seen as whiter-than-normal areas in this portion of the brain, as shown in Fig. 56.1. In addition to these more obvious abnormalities, a systematic analysis of MR images shows that the T1 signal abnormality is widespread and found in the limbic and extrapyramidal systems, and generally throughout the white matter. A generalized shortening of the T2 signal also occurs. This abnormality is less evident on visual inspection of the images because of the generally short duration of T2 signals. These abnormalities have been linked to an increase in the cerebral manganese content. The abnormalities become more prominent with time and regress after successful liver transplantation. The unexpected finding of high T1 signals in the pallidum should suggest the possibility of liver disease.
Cerebral Blood Flow and Glucose Metabolism
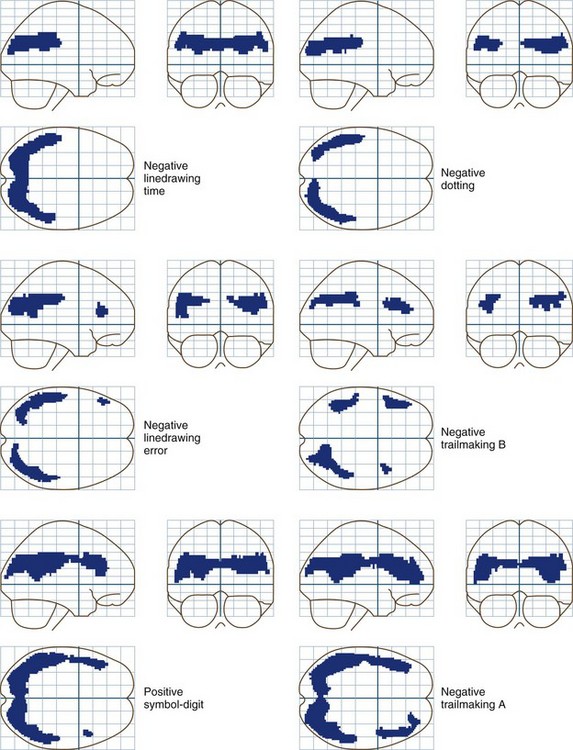
Whole-brain measurements of cerebral blood flow (CBF) and metabolism are normal in patients with grade 0 to 1 HE. Reductions occur in more severely affected patients. Sophisticated statistical techniques designed to analyze images have made it possible to identify specific brain regions in which glucose metabolism is abnormal in patients with low-grade encephalopathy and abnormal neuropsychological test scores (Lockwood et al., 2002). These positron emission tomography (PET) data show clearly that minimal forms of HE are caused by the selective impairment of specific neural systems rather than global cerebral dysfunction. Reductions occur in the cingulate gyrus, an important element in the attentional system of the brain, and in frontal and parietal association cortices. These PET data are in accord with cortical localizations based on the results of neuropsychological tests. Fig. 56.2 shows the results of correlation analyses between scores on selected neuropsychological tests and sites of reduced cerebral glucose metabolism.
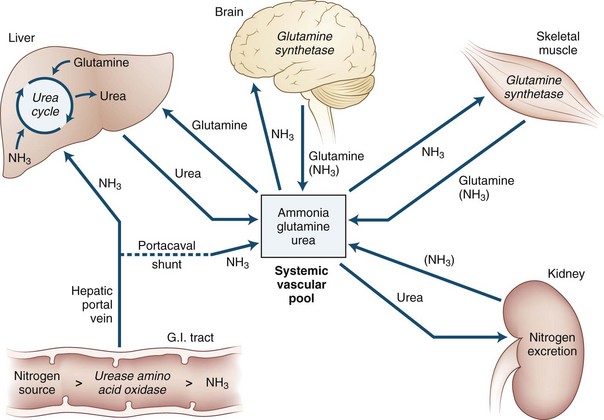
Role of Ammonia
Ammonia is always extracted by the brain as arterial blood passes through the cerebral capillaries. When ammonia enters the brain, metabolic trapping reactions convert free ammonia into metabolites (Fig. 56.3). The adenosine triphosphate (ATP)-catalyzed glutamine synthetase reaction is the most important of these reactions. The blood-brain barrier is approximately 200 times more permeable to uncharged ammonia gas (NH3) than it is to the ammonium ion (NH4+); however, because the ionic form is much more abundant than the gas at physiological pH values, substantial amounts of both species appear to cross the blood-brain barrier. Because of this permeability difference and because ammonia is a weak base, relatively small changes in the pH of blood relative to the brain have a significant effect on brain ammonia extraction. As blood becomes more alkalotic, more ammonia is present as the gas and cerebral ammonia extraction increases; however, the role this has in the production of HE is not known. The permeability surface-area (PS) product of the blood-brain barrier may be affected by prolonged liver disease. However, the experimental data about this change are in conflict: one study reported an increase in the PS product, and another reported no change. An increased PS product could explain in part the toxin hypersensitivity that develops with time.
Neuropathology
The Alzheimer type II astrocyte is the neuropathological hallmark of hepatic coma. An account of the original descriptions of this change was provided in translation by Adams and Foley in 1953. In this report, they presented their own findings of this astrocyte change in the cerebral cortex and the lenticular, lateral thalamic, dentate, and red nuclei, offering the tentative proposal that the severity of these changes might be correlated with the length of coma. The cause of the astrocyte change was established by studies that reproduced the clinical and pathological characteristics of HE in primates by continuous infusions of ammonia. In studies of rats with portacaval shunts, astrocyte changes become evident after the fifth week. Before coma develops, astrocytic protoplasm increases and endoplasmic reticulum and mitochondria proliferate, suggesting that these are metabolically activated cells. After the production of coma, the more typical signs of the Alzheimer type II change became evident as mitochondrial and nuclear degeneration appeared. Norenberg (2007) suggested that HE is an astrocytic disease, although oligodendroglial cells are affected as well. More recent evidence from his laboratory has shown that ammonia affects a wide variety of astrocytic functions and aquaporin-4.
Treatment
Ideally, the management of cirrhosis should involve a cooperative effort between hepatologists, surgeons, neurologists, and psychologists, with additional input from nurses and dieticians. Practice guidelines published by the American College of Gastroenterology identify four goals: (1) provide supportive care, (2) identify and treat precipitating factors, (3) reduce the nitrogenous load from the gut, and (4) assess need for long-term therapy (Blei and Cordoba, 2001; Ferenci et al., 2002).
Lactulose
Lactulose is a mainstay for the treatment of both acute and chronic forms of HE. Its utility in the secondary prevention of HE was supported by a recent open-label placebo-controlled study of patients who had recovered from an initial episode of HE (Sharma et al., 2009). In the lactulose-treated group, 19.6% developed recurrent HE during a 1- to 14-month follow-up compared to 46.8% in the placebo group (P = 0.02). Lactulose is a synthetic disaccharide metabolized by colonic bacteria to produce acid and causes an osmotic diarrhea. A widely held but incorrect theory concerning the mechanism of action of lactulose centers on its ability to acidify the colon. Acidification presumably trapped ammonia as the charged and nonabsorbable ammonium ion, thereby preventing ammonia absorption. This theory has been questioned because lactulose treatment does not increase the fecal ammonia concentration or the total amount of ammonia excreted. The effect of lactulose is attributable to its role as a substrate in bacterial metabolism, leading to an assimilation of ammonia by bacteria or reducing deamination of nitrogenous compounds. It is probably the single most important agent in the treatment of acute and chronic encephalopathy. The usual dose of lactulose is 20 to 30 g, 3 or 4 times a day, or an amount sufficient to produce 2 or 3 stools per day. Lactulose also can be given as an enema. Lactitol, another synthetic disaccharide, is also effective. Although it is not yet available in the United States, it may have some advantages over lactulose because it can be prepared in a crystalline form that may make it more acceptable to patients who may object to the taste of lactulose preparations.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


