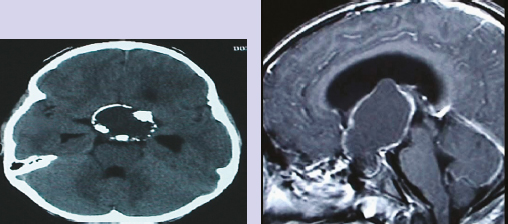
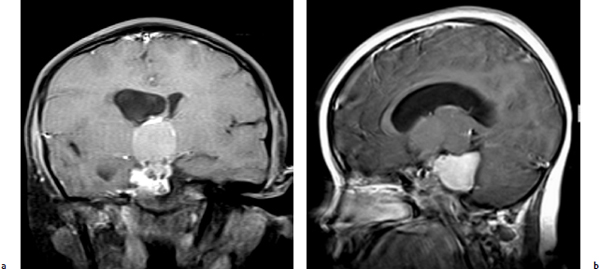
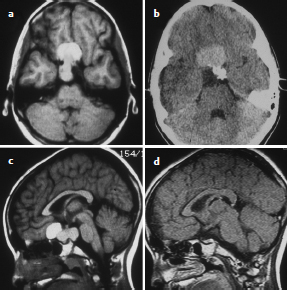
Chapter 9 Case A 14-year-old girl came to medical attention with diabetes insipidus and a visual field defect. Participants Subtotal Resection with Focal Irradiation for Craniopharyngiomas: Frederick A. Boop and Scott D. Wait Radical Resection of Pediatric Craniopharyngiomas: Jeffrey H. Wiskoff and Robert E. Elliott Moderators: Total Removal vs. Subtotal Removal with Radiation for Craniopharyngiomas: Edward R. Smith and R. Michael Scott Here is a letter I recently received from the mother of a child with a craniopharyngioma: I have a 15-year-old son who was diagnosed at the age of 10 with a golf ball–sized craniopharyngioma. He had a gross total resection. … He has all the usual complications: panhypopituitarism, diabetes insipidus, hyperphagia, hypothalamic obesity, and visual loss, plus he suffered a stroke when the surgeon “nicked” an artery. He also developed severe behavioral problems. He is extremely violent and self-abusive. One day, we noticed he didn’t have any vision at all. We took him to his ophthalmologist and she recommended we take him to see his neurologist because she couldn’t see anything wrong. We had appointments with endocrinology too so his neurologist worked him in. He was admitted and had an emergency MRI. It was found that a herpes virus had attacked his retina. His neurologist told us the MRI had shown a recurrence of the craniopharyngioma in the sella region. He gave us three options: (1) wait and see and re-scan in 3 months, (2) stereotactic radiosurgery, or (3) do nothing. The final option was given only because our son had developed other issues such as hypertension, spinal fusion due to idiopathic scoliosis, dystonia, two Staph. aureus infections, sleep apnea, diabetes, enlarged liver with elevated liver enzymes, lung depletion in the lower quadrants of his lungs, and depression. Right now we are planning on doing the radiation in the near future. This letter is not to be misconstrued as an indictment of surgery for craniopharyngiomas but rather as an example of what is likely when a less experienced team cares for a child with a complicated craniopharyngioma. When one stops to consider that craniopharyngiomas represent only 1 to 2% of pediatric brain tumors, that the average children’s hospital may see 30 to 40 pediatric tumors per year, and that there are generally two or more neurosurgeons on staff at a given hospital, then the average pediatric neurosurgeon might operate on only one craniopharyngioma every 2 to 3 years.1 Thus, outside of major referral centers, it would be difficult for most neurosurgeons to gain enough experience with these tumors to consider themselves experts. More importantly, in interpreting the published literature on surgery for craniopharyngiomas, one cannot extrapolate the outcome or morbidity from a Wisoff, Yasargil, or Al-Mefty to one’s own experience.2 The care of children with these tumors is complex, requiring a well-organized multidisciplinary team willing to consider various treatment options for each individual patient. The team must provide a careful long-term follow-up to optimize functional recovery. The treatment recommendations for the child cannot simply be “take it all out” versus “treat with focal irradiation.” The reason that there are so many reported treatment options for this disease, such as observation, intracystic therapy, endoscopic drainage, stereotactic radiosurgery, conformal radiotherapy, proton beam therapy, and limited to complete resection, is that each of these treatments works well for some patients and none of these treatments works best for all patients. The treatment considerations for the child who presents with a headache, short stature, and normal vision with a small sellar lesion should be different from that for the child who presents with obtundation, panhypopituitarism, and acute hydro-cephalus secondary to a giant craniopharyngioma3 (Figs. 9.1, 9.2). The preoperative evaluation of each child is essentially the same. It includes high-definition magnetic resonance imaging (MRI), a formal visual evaluation, a detailed endocrinologic workup including stimulation testing, and psycho social evaluation including developmental history, school performance, family stability, and available community support.4 In the past quarter of a century, our microsurgical skills and training have allowed us to move from reporting morbidity and mortality throughout the 1970s to reporting basic functional outcome measures, such as the Glasgow Outcomes Scale at the end of the last century, and then to more detailed functional scales such as those of Karnofsky, Lansky, and others. In the current era, reporting of outcomes from craniopharyngioma treatment has moved to detailed neuropsychometric studies, quality-of-life measures, evaluations of school performance including attentional and processing problems in addition to visual acuity and fields, details of endocrinopathy and vasculopathy, and measures of progressive disease. Computed tomography (CT) and magnetic resonance angiography give us accurate assessments of postoperative pseudoaneurysms, moyamoya disease, and small-vessel arteritis. We also know that with each major intervention for craniopharyngiomas—surgery, radiotherapy, cyst catheter replacement, or shunt revision—the child will lose some measure of function. So when we review a series such as that reported by Hoffman, in which only 60% of children had a gross total resection of their tumor, and of those 15 to 20% recurred over time, the majority of these children will require a second craniotomy for recurrent disease, often followed by irradiation.5 Under these circumstances, for the child who has had two or three major craniotomies with or without a shunt or shunt revision, followed by medial forebrain irradiation, it is not realistic to expect the child to remain highly functional. Thus, minimizing the number of potential interventions is critical when designing a treatment strategy for these children.6 A child with a small suprasellar craniopharyngioma should have an 80% chance for long-term cure with either complete resection of the tumor or with focal irradiation. The long-term recurrence risk after either treatment is approximately 20%. After focal irradiation, the child will likely eventually require a replacement of growth hormone, cortisol, and thyroid hormones, but is not likely to develop diabetes insipidus. The child having had surgical extirpation will likely require treatment for diabetes insipidus in addition to the other three hormones. Certainly, hormone replacement for diabetes insipidus is easier now that desmopressin is in a pill form, but in an unreliable social setting or for the family who cannot afford the medications, diabetes insipidus is life-threatening.7 Fig. 9.1a,b (a) This is a T1-weighted gadolinium enhanced coronal image of an 8-year-old female presenting with headaches, normal visual exam, and normal endocrine status. Note the supracellar tumor extending into the third ventricle and causing hydrocephalus. (b) A sagittal T1 gadolinium enhanced image of the same patient showing extension of the tumor into the posterior fossa nearly to the foramen magnum. Fig. 9.2a,b The third ventricular portion in the same patient as in Fig. 9.1 was resected through a transcallosal approach and the posterior fossa portion through a retrosigmoid approach, leaving tumor stuck to the hypothalamus. The patient then received focal conformal radiation to the small residual tumor. Two years following surgery, she is currently free of headaches with normal vision and requires only the replacement of growth hormone. Regarding diabetes insipidus, many surgical articles have discussed the importance of preserving the pituitary stalk, even though most of the time the patient will develop diabetes insipidus after resection of the craniopharyngioma. In the case of larger craniopharyngiomas, diabetes insipidus may be secondary to hypothalamic injury, not just the pituitary stalk effect. Thus, if one considers diabetes insipidus as a dependent variable, children with this disease have an increased risk of obesity, attentional problems, and learning difficulties compared with those who do not have diabetes insipidus.8 Given that diabetes insipidus is seen in association with radical resection of craniopharyngiomas in up to 90% of patients, whereas it is an uncommon consequence of treating a child with focal irradiation, one can understand that larger craniopharyngiomas with hypothalamic involvement would be better suited to partial resection and focal radiotherapy. These data have led many neurosurgeons to propose grading systems for craniopharyngiomas in which treatment decisions are based on preoperative imaging characteristics. Puget and colleagues,9 for example, have proposed a three-tier system in which children with a craniopharyngioma but no hypothalamic involvement undergo surgery with curative intent, whereas those with significant hypothalamic involvement undergo partial resection to decompress neurologic structures and reduce the treatment volume required for postoperative radiosurgery. The optimal treatment of a child presenting with a craniopharyngioma is complex and should be based on a multidisciplinary evaluation when possible. Given the number of treatment options, the treatment should take into account the experience of the treating surgeon and radiation oncologist, the extent of involvement of neurovascular structures seen on imaging, and the resources available to the patient and family. Excellent long-term results can be obtained in the hands of experienced surgeons as long as neuroendocrine support and long-term follow-up are provided. Similar results can be obtained by less experienced teams with limited surgery and focal irradiation. In this group of children, progression-free survival rates of 80% or better can be expected at 10 and 20 years, with only a third of the patients requiring lifelong treatment for diabetes insipidus.7 Using a paradigm of subtotal resection and irradiation should virtually eliminate surgical deaths. Thus, for the majority of pediatric neurosurgery centers and for children with a less reliable social milieu, this approach allows for a better outcome with less up-front risk.10 For children with a recurrent craniopharyngioma or progression of disease after prior incomplete resection, it is best to consider referral to a specialty center for further treatment when possible. We are currently conducting a detailed prospective study to determine whether proton beam radiotherapy can improve the quality of life in children with this disease [A Phase II Trial of Limited Surgery and Proton Therapy for Craniopharyngioma and Observation for Craniopharyngioma After Radical Resection (NCT01419067); http://clinicaltrials.gov/ct2/show/NCT01419067.] We published a monograph in 1996 on the management of craniopharyngiomas, in which we listed 10 recommendations.2 Based on the improvements in care and in the understanding of this disease since that time, we have modified those 10 recommendations as follows: 1. The goal of treating craniopharyngiomas should be to control tumor growth while preserving the patient’s cognitive, visual, and endocrine function. 2. Microsurgical decompression should be the initial treatment in any patient with rapid visual or neurologic decline or obstructive hydrocephalus. For hydrocephalus secondary to a large cystic craniopharyngioma, stereotactic or endoscopic placement of an Ommaya reservoir into the cyst, followed by aspiration and irradiation, has proven to be an effective alternative if the hydrocephalus is alleviated. 3. Microsurgical resection should be the initial consideration in children younger than 5 years of age in an effort to avoid the potential deleterious effects of radiation on the developing brain. 4. For patients with a large solid component to their tumor, both complete resection with curative intent and subtotal resection followed by focal irradiation appear to have equivalent control rates. Subtotal resection with focal irradiation affords better preservation of hypothalamic function. 5. Transsphenoidal resection should be considered the initial treatment for tumors located primarily within the sella turcica. 6. Surgical removal should be considered only for patients who have ready access to appropriate endocrinologic follow-up and replacement hormones, and for whom the family is capable of reliably providing for postoperative endocrinologic needs. Patients returning to underdeveloped countries or rural areas are at increased risk of death if these needs are not considered preoperatively. Compared with surgery, radiation therapy clearly reduces the likelihood of diabetes insipidus and the need for multiple hormone replacement. 7. For patients with primarily cystic tumors, intracystic therapy with the radioisotopes of phosphorus (32P) or yttrium (90Y) or with interferon appears to be efficacious, but its long-term ability to control the tumors has been called into question and many centers, including ours, have stopped using this modality in children. 8. Patients in whom a complete resection has been attempted by a skilled microneurosurgeon but has failed because of the tumor’s adherence to vital structures may be closely followed with neuroimaging if progression is not likely to compromise visual or neurologic function. As two thirds of cases progress within 3 years of surgery, focused radiosurgery should be done at the time of clinical or radiographic progression. It should not be done for a fleck of residual calcification seen on a postoperative computed tomogram. 10. Stereotactic radiosurgery is best reserved for solid components of craniopharyngiomas that are small (less than 20 mm in diameter) and separated from the optic apparatus by 3 to 5 mm or more. Craniopharyngiomas comprise roughly 3% of all intracranial neoplasms11,12 and are the most common nonglial brain tumor of childhood, constituting 6 to 8% of all pediatric brain tumors.13,14 On a population scale, however, they are relatively rare lesions with an incidence of only 0.13 per 100,000 person years.15 Fewer than 350 combined adult and pediatric craniopharyngiomas are diagnosed each year in the United States, and less than half of these occur in children.15,16 Thought to arise from embryological remnants of the craniopharyngeal duct, these benign epithelial neoplasms with solid, cystic, and calcified components can arise anywhere along an axis from the third ventricle to the pituitary gland.17–21 The benign histology of craniopharyngiomas, however, belies their rather malignant clinical course in children. Described by Harvey Cushing22 as “one of the most baffling problems to the neurosurgeon,” their close proximity to the visual apparatus, circle of Willis, pituitary stalk, and hypothalamus predisposes these patients to severe adverse sequelae both at presentation and after treatment. Common findings include headache, vision loss, diabetes insipidus, panhypopituitarism, short stature, hypothalamic dysfunction with behavioral and memory disturbances, hyperphagia, and obesity. Debate persists regarding the optimal treatment of patients with craniopharyngiomas. Regardless of the strategy selected, however, definitive tumor control or cure should be the goal of any treatment for pediatric craniopharyngiomas. Two critical factors for a potential cure are the extent of surgical excision and cranial irradiation. Some centers advocate radical resection for surgical cure, whereas others favor limited resection followed by radiation therapy to potentially limit injury to the hypothalamus. Both major paradigms provide similar rates of disease control and overall survival.23–39 Although radical resection may have a higher potential for immediate perioperative morbidity,9,23,29,40–44 limited resection and radiation therapy causes more delayed morbidity including panhypopituitarism, visual deterioration, cognitive and attentional dysfunction, secondary central nervous system neoplasms, and cerebrovasculopathy, namely moyamoya disease.31,45–53 Palliative procedures such as stereotactic cyst aspiration or Ommaya reservoir drainage may provide relief from the compression of neural and visual structures, but these effects are invariably transient. Progressive solid and cystic tumor recurrence and growth is inevitable. We believe such therapies should not be considered definitive or adequate treatment early in the course of disease. The relative scarcity of craniopharyngiomas, the persistent lack of consensus regarding optimal treatment, and the potential morbidity of all forms of treatment combine to make it difficult, if not impossible, to determine the best management strategy. Given similar rates of disease control and survival with the two main treatment strategies, the focus of outcome assessment has shifted to quality-of-life metrics.9,31,54–58 However, detailed quality-of-life outcomes from large series of uniformly treated patients are scarce. In this section of the chapter, we describe our preferred treatment paradigm for craniopharyngiomas in children: radical resection with the aim of surgical cure. Depending on the patient’s age and the clinical status before surgery, we prefer a complete evaluation by various specialists that includes ophthalmologic, endocrinologic, and neuropsychological testing. Parents and families are counseled as to the expected short- and long-term postoperative course. Our preoperative imaging protocol consists of MRI with frameless stereotactic image acquisition and CT. CT provides detailed information about the extent and location of tumoral calcification. Careful evaluation of multiplanar MRI scans is essential to understand the often complex relationship that craniopharyngiomas have to the visual apparatus, hypothalamus, and surrounding vasculature, and will lead to improved outcomes. First, the location of the tumor in relation to the optic apparatus must be determined. Tumors can be entirely subchiasmatic primarily within the sella, prechiasmatic with or without a subfrontal extension, retrochiasmatic involving the floor of the third ventricle and the hypothalamus, or have a complex relationship to the chiasm with both prechiasmatic and retrochiasmatic components. Second, attention must be paid to the relationship of the dorsal aspect of the tumor and the hypothalamus. An increased involvement and deformation of the hypothalamus has been shown to predict the level of preoperative hypothalamic dysfunction as well as the operative morbidity of resection. Third, as craniopharyngiomas enlarge, they can form multi-lobulated cysts that extend along the pathways of least resistance and invade nearby anatomic spaces in the anterior, middle, and posterior fossae. These extensions must be recognized to optimize the surgical approach and minimize any retraction injury to normal brain parenchyma. Given the variability of the precise location and size of craniopharyngiomas, a variety of approaches have been described by different surgeons. These include the sub-frontal,5,13,59–62 pterional,23,26,44,63,64 combined,9,24,37,39,41 bi-frontal interhemispheric,65,66 transcallosal,67 subtemporal,68 transpetrosal,69 and transsphenoidal approaches.70–73 We prefer a modified pterional exposure that includes removal of the supraorbital rim, anterior orbital roof, and the zygomatic process of the frontal bone. This approach provides the shortest, most direct route to the suprasellar region. It minimizes frontal and temporal lobe retraction with wide splitting of the sylvian fissure, allows early release of cerebrospinal fluid from the sylvian and carotid cisterns to aid brain relaxation, and facilitates early visualization of the carotid arteries and optic apparatus. Tumors extending from the pontomedullary junction to above the foramen of Monro can be successfully and safely removed through this approach without the need for corticectomy or sacrifice of the olfactory nerve, or potential cognitive dysfunction from the retraction of both frontal lobes. Surgical adjuncts include the Cavitron Ultrasonic Surgical Aspirator (CUSA; Tyco Healthcare Radionics, Burlington, MA), frameless stereotaxy, and rigid and flexible endoscopes, and these should all be used when appropriate. Recently, we have found that endoscopic visualization during dissection of the tumor from the ventral surface of the chiasm and floor of the third ventricle greatly enhances the safety of tumor removal in this critical region and enables the complete removal of small fragments of tumor, calcium deposits, or both, that may or may not contain viable tumor cells. The endoscope is also useful for intraventricular visualization and the potential resection of tumor that lies within the third or lateral ventricles and is not accessible through the transsylvian approach. We reserve the transsphenoidal approach for tumors that are primarily or completely within the sella turcica. After intubation and the induction of anesthesia, the patient is positioned and stereotaxy is registered. Then dexamethasone (0.1 mg/kg), phenytoin (15 mg/kg), and cephalexin (25 mg/kg) are administered. Mannitol (0.25 g/kg) is given at the time of skin incision to aid brain relaxation. The diuretic effect is maximal within 1 hour of infusion and, ideally, at the time of brain and tumor manipulation. Hyperventilation and the progressive drainage of cerebrospinal fluid from the sylvian and basal cisterns usually provide excellent brain relaxation, even in the presence of hydrocephalus. Ventricular drainage is reserved for cases refractory to these maneuvers or in patients with severe, increased intracranial pressure unresponsive to medical management. However, if severe hydrocephalus is present or if there is a significant solid tumor component superiorly within the third ventricle, a 4-mm endoscope is placed into the lateral ventricle and held in place with a rigid retractor. This maneuver allows the surgeon to alternate visualization and the dissection of tumor from the endoscopic, intraventricular, or microscopic transsylvian routes. A Z-plasty skin incision posterior to the hairline is made from the tragus to just beyond the midline. The temporalis fascia and muscle are sharply incised with a No. 15 blade and bluntly dissected from the underlying calvaria with a periosteal elevator to allow for excellent reapproximation at the end of the procedure and to minimize atrophy of the temporalis muscle. A one-piece, modified pterional craniotomy with removal of the anterior orbital roof, supraorbital rim, and zygomatic process of the frontal bone is then performed with the craniotome and a chisel and mallet. A brain retractor is used to prevent injury to the orbital contents or laceration of the periorbita during the orbital and supraorbital osteotomies. The dura is dissected from the sphenoid bone, which is removed with rongeurs down to the supraorbital fissure. The dura is then elevated with a dural hook and incised in a C-shaped fashion. Especially for large tumors that distort the anatomy of or extend beyond the suprasellar cisterns, identifying the vascular anatomy provides critical internal landmarks for safe navigation. Laterally, the sylvian fissure is widely split and the branches of the middle cerebral artery are identified. Arachnoidal dissection of the fissure proceeds medially to the bifurcation of the internal cerebral artery. Once the carotid artery comes into view, the anterior cerebral artery and optic nerve, chiasm, and tracts are carefully inspected to reveal the relationship of these structures to the tumor (Figs. 9.3, 9.4). We caution against early decompression of the cystic portion of the tumor, as this can result in redundancy of the tumor capsule and the overlying attenuated arachnoid. This loss of turgor can obscure the planes of dissection. The overarching strategy for craniopharyngioma resection is to develop an arachnoid plane circumferentially around the tumor within the suprasellar cisterns followed by inspection and possible sectioning of the stalk. The last and most critical step is manipulation and excision of the dorsal portion of the tumor involving the hypothalamus. Fig. 9.3a–d A 9-year-old girl presented with a severe, progressive headache. On examination, she was found to have partial palsy of the third cranial nerve on the left and 20/40 visual acuity on the left. (a,c) Magnetic resonance imaging (MRI) scans after gadolinium enhancement revealed a 4-cm mixed cystic and solid tumor with a post-fixed chiasm. (b) Solid calcification in the left suprasellar region was apparent on the computed tomography (CT) scan. (d) Through a right pterional craniotomy, the patient underwent gross total resection of the adamantinomatous craniopharyngioma with transient worsening but eventual improvement in her cranial nerve palsy. Her visual acuity improved to 20/25 after surgery. Although the pituitary stalk was preserved, she developed diabetes insipidus and requires desmopressin. She is now 18 years past surgery, has been without disease recurrence, and completed graduate school after attending college. Working in the opticocarotid, prechiasmatic, and carotidotentorial triangles, an arachnoidal plane is developed between the tumor capsule and the arteries of the circle of Willis. Careful attention must be paid to ensure preservation of the basal perforators. This plane is developed in a posterior direction until the basilar artery is identified through the usually intact membrane of Liliequist. This extracapsular dissection is usually facilitated by well- demarcated and preserved arachnoidal planes. In patients with recurrent tumors, these planes can be heavily scarred and may require an increased use of sharp microdissection. After the cerebral vasculature is separated from the tumor capsule, the cyst can be aspirated and the solid components debulked. All attempts should be made to preserve the capsule of the tumor to allow gentle traction for eventual dissection of the tumor from its remaining attachments, especially the floor of the hypothalamus. With continuing respect for the arachnoidal planes, the surgeon then dissects the tumor free from the optic chiasm and the contra-lateral carotid artery and its branches. Although an attempt is always made to identify and preserve the pituitary stalk, we have found this successful in only 30% of patients. We recommend sectioning the stalk as distal as possible, without compromising negative margins, to potentially limit the severity of diabetes insipidus. After the tumor is separated from the entire circle of Willis and the pituitary stalk and optic apparatus, the capsule is grasped, and, with a combination of gentle traction and blunt dissection, a gliotic plane is developed between the dorsal aspect of the tumor and the floor of the third ventricle and hypothalamus in the region of the tuber cinereum. After the tumor is removed, the entire tumor bed must be inspected for residual disease with either a micro-mirror or an angled endoscope. Papaverine-soaked Gelfoam pledgets are then placed around the arteries of the circle of Willis to help ameliorate vasospasm (Fig. 9.4) and are removed before dural closure. Fig. 9.4a,b (a) This intraoperative photograph taken after splitting of the right sylvian fissure confirmed the prechiasmatic nature of the craniopharyngioma seen in Fig. 9.3. The left optic nerve is elevated by the tumor, rotated into view, and exhibits pallor. (b) After gross total resection of the tumor, the optic nerves were decompressed and vasospasm was evident in the right internal carotid artery and the A1 segment of the anterior cerebral artery. If the tumor has a significant retrochiasmatic or intraventricular component, the lamina terminalis must be fenestrated (Fig. 9.5). The lamina terminalis is distinguished from the chiasm by its pale, avascular appearance and is often distended and attenuated by the underlying tumor. Tumor within the third ventricle can be simultaneously delivered through both the lamina terminalis and from below the chiasm. We find the use of a 4-mm endoscope inserted into the third or lateral ventricle to be extremely helpful to assist in the delivery of the intraventricular component of the tumor, obviating the need for a transcallosal approach to achieve complete resection. For tumors with a significant extension into the sella turcica, removal of the tuberculum sellae and posterior planum sphenoidale may be necessary to gain adequate exposure of the intrasellar space. After tumor removal, all bony defects in the sinuses must be repaired to prevent cerebrospinal fluid fistulas. Fig. 9.5a–d A 7-year-old boy presented with headache and behavioral outbursts. (a,b) MRI scans revealed a 5-cm retrochiasmatic tumor with significant extension into the third ventricle, causing obstructive hydrocephalus. (c,d) After gross total resection of his adamantinomatous craniopharyngioma through a right pterional approach and fenestration of the lamina terminalis, he remained neurologically, visually, and hormonally intact, and his hydrocephalus resolved after the tumor was removed. He did, however, experience slight worsening of his short-term memory but was able to do well in school and currently attends college. He remains free of disease 14 years after resection. Following surgery and neurologic examination, all children are transferred immediately to the pediatric intensive care unit. A multidisciplinary team of pediatric endocrinologists, neuro-oncologists, and intensivists collaborate in the postoperative care. Frequent urine and electrolyte analyses are done to monitor for and aggressively treat electrolyte disturbances, namely diabetes insipidus. Dexamethasone is tapered over the course of 1 week, and phenytoin is continued for 3 weeks after surgery. The phenytoin is further continued for extended periods only in patients with seizures that cannot be attributed to electrolyte disturbances. Postoperative MRI and CT scans are done within 48 hours after surgery to ensure complete resection. Surveillance MRI scans and clinical follow-up occur every 3 months during the first year, every 4 months during the second year, every 6 months for the next 3 years, and every year for the next 5 years. Frequent imaging allows for the early detection of recurrence while tumors are small and preferably asymptomatic. However, long-term imaging and follow-up are important, as late recurrences have been reported. Regular evaluations by dedicated pediatric endocrinologists, ophthalmologists, and neuro-oncologists are essential in managing these cases long-term. In the era of MRI, radiographically confirmed complete resection is possible in 80 to 100% of patients. Perioperative mortality after aggressive surgery has also declined substantially over the past two decades secondary to advances in neuroimaging and microsurgical techniques, from more than 10% to 0 to 4% in most current series.5,9,13,23–26,29,30,32,39,40,45,63,64,74–80 Multiple authors report that the surgeon’s experience with craniopharyngiomas has a significant impact on the likelihood of achieving complete resection and good functional outcomes.9,35 Surgeons performing more than two operations per year for radical resection had good outcomes in 87% of children compared with only 52% in those performing fewer operations.35 Numerous centers have reported excellent rates of disease control and functional outcomes with the strategy of radical resection. In a large series by Zuccaro,39 complete resection was achieved in 69% of 153 children. All children who underwent complete resection were in school and no more than 1 year behind in grade level, in contrast to only 62% of children who had limited resection and radiation. Di Rocco and colleagues25 reported complete resection in 78% of 54 children treated with curative surgical intent. In their series, overall improvement in the patients’ intelligence quotient (IQ) occurred after resection with a mean postoperative IQ of 112 (range 95–130). All but two of 50 surviving patients enjoyed normal social interactions. In a series by Hoffman and associates,5 26 of 27 children who underwent aggressive resection had IQ scores at or above average levels. Although 16 children had memory deficits, 14 of them attended regular schools. These authors contend that “memory impairment did not interfere with school progress if intelligence was adequate.” Yaşargil and colleagues44 reported good outcomes in 72.5% of children after initial surgery, and Fahlbusch and associates63 reported functional independence in 78% of adult and pediatric patients after radical resection. In our series of 86 children who underwent radical resection of a craniopharyngioma, gross total resection was accomplished in all 57 patients with primary tumors (100%) and in 18 of 29 (62%) with recurrent tumors with acceptably low morbidity (Table 9.1). In contrast to the findings of other centers of increased morbidity, mortality, and worse functional outcomes at reoperation,23,29,30,44,63,81–85 we found no such differences in our series. Good to excellent functional outcomes were achieved in 80% of children, and more than 60% of college-aged patients either attended or graduated from college—a clear indication of the high functionality of the majority of these children. New hypothalamic morbidity occurred in 25% of children and was mild or moderate in all but one case. Fewer than 20% of our patients developed obesity, and only two patients developed severe or morbid obesity. These results contrast greatly with those from a German multicenter study that reported severe obesity in 44% of 185 children treated for craniopharyngiomas using various treatment modalities.56 Although some centers contend that an increasing tumor size limits the extent of resection and local disease control,38,40,63,86–92 we agree with other authors that size has no impact on the ability to achieve gross-total resection, at least for the initial tumors.39,60,64,93 Nevertheless, given the large size and multicompartmental nature of giant craniopharyngiomas, a flexible and at times staged approach may be required for successful and safe extirpation of these tumors (Fig. 9.6). Table 9.1 Morbidity and Mortality in 86 Children after Radical Resection of Craniopharyngiomas
Treatment of Craniopharyngiomas: Total Removal vs. Subtotal Removal with Radiation
Subtotal Resection with Focal Irradiation for Craniopharyngiomas
Preoperative Evaluation
Treatment Strategies
Management of Craniopharyngiomas
Radical Resection of Pediatric Craniopharyngiomas
Treatment Philosophy
Preoperative Evaluation
Surgical Approaches
Operative Technique
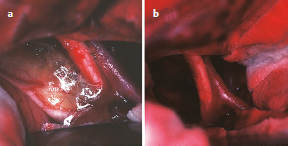
Postoperative Care
Outcomes and Complications
No. of Patients (%) | ||
Primary | Recurrent | |
Perioperative mortality | 2 (3.5%) | 1 (3.4%) |
Neurologic morbidity* | ||
Stroke | 2 (4%) | 2 (9%) |
Mild hemiparesis | 1 (2%) | 0 (0%) |
Transient cranial nerve palsy | 8 (16%) | 1 (4%) |
Permanent cranial nerve palsy | 1 (2%) | 1 (4%) |
Lethargy/abulia | 2 (4%) | 1 (4%) |
Visual acuity | ||
Preoperative deficit | 14 (27%) | 15 (60%) |
Improved | 10 (19%) | 3 (12%) |
Stable | 35 (67%) | 17 (68%) |
Worse | 7 (13%) | 5 (20%) |
Visual fields | ||
Preoperative deficit | 23 (43%) | 22 (85%) |
Improved | 13 (25.5%) | 7 (27%) |
Stable | 25 (49%) | 12 (46%) |
Worse | 13 (25.5%) | 7 (27%) |
Diabetes insipidus | ||
Preoperative | 6 (12%) | 19 (73%) |
Postoperative, new | 33 (73%) | 6 (67%) |
Postoperative, total | 39 (78%) | 25 (89%) |
Anterior pituitary dysfunction: mean number of hormones required ± SD | 2.5 ± 1.1 | 2.1 ± 0.9 |
Abbreviations: SD, standard deviation.
Although our data did corroborate prior studies reporting worse overall survival for children with recurrent tumors,34,38,44,63,81,94 subgroup analysis revealed excellent survival for children with nonirradiated recurrent tumors and those of smaller size at reoperation. Thus, prior failed radiation therapy and a large size at recurrence significantly limited our ability to achieve complete resection, the only remaining option for potential cure for these patients, and resulted in worse overall survival. In contrast, prior radical resection per se did not diminish the chance of achieving complete resection at reoperation, leading to improved disease control and survival. Fourteen children experienced a total of 15 recurrences after gross total resection at our hospital. All underwent reoperation at the time of recurrence, except one child who had radiosurgery because of fusiform dilation of the internal carotid artery. Gross total resection was achieved in 79% with no surgical morbidity or mortality. One patient had slight deterioration in vision but none of the children experienced hypothalamic or memory dysfunction. Overall survival for this cohort was 92% at a mean follow-up of 8 years, markedly higher than the rate of survival in patients with recurrent tumors reported in the literature.
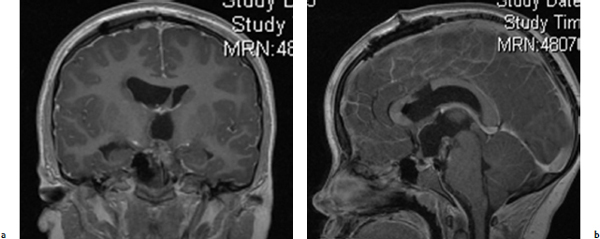
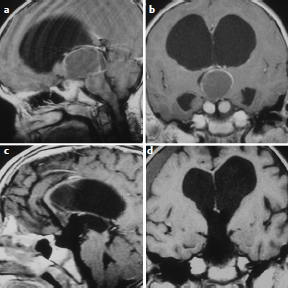
Fig. 9.6a–c (a,b) A 12-year-old boy presented with headache and was found on CT to have a giant, mixed cystic and solid tumor with extensive calcification in the suprasellar region and cystic extension into the left middle fossa. (c) After gross total removal of this adamantinomatous craniopharyngioma, he experienced a stroke that left him with a mild right hemiparesis, which improved over time. He did not develop diabetes insipidus but was left with a new right superior quadrantanopsia, likely from retraction of the left side of the optic chiasm during tumor removal. He experienced a 2-cm recurrence that was also removed 5 months after his initial surgery. He received passing grades at appropriate levels in school, required no adjuvant therapy, and has been free of disease for 23 years since his last surgery.
Related posts:
 Management of Symptomatic Carotid Stenosis
Management of Symptomatic Carotid Stenosis
 Surgical Removal of Tuberculum Sellae Meningiomas: Endoscopic vs. Microscopic
Surgical Removal of Tuberculum Sellae Meningiomas: Endoscopic vs. Microscopic
 The Application of Artificial Discs
The Application of Artificial Discs
 Management of Trigeminal Schwannoma: Microsurgical Removal vs. Radiosurgery
Management of Trigeminal Schwannoma: Microsurgical Removal vs. Radiosurgery
 Treatment of an Arteriovenous Malformation in Eloquent Areas
Treatment of an Arteriovenous Malformation in Eloquent Areas
 Resection of Gliomas in Eloquent Areas of the Brain
Resection of Gliomas in Eloquent Areas of the Brain



