Chapter 83 Tumors of the Brain and Spine
Introduction
Primary central nervous system tumors comprise, in total, the second most common form of childhood cancer, exceeded only by the leukemias [CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007a; Preston-Martin, 1996; Ries et al., 1997]. Although relatively rare compared to other pediatric neurological conditions, these neoplasms cause significant neurologic morbidity and mortality. Unlike the situation in adults, where metastatic brain tumors outnumber primary central nervous system neoplasms by a ratio as high as 10 to 1, in the pediatric years, metastatic lesions are much less common and primary brain and spine tumors predominate [Bleyer, 1999; CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007b; Ries et al., 1997; Smith et al., 1998].
Tumors of the brain and spine constitute approximately one-quarter of all childhood cancers. In the United States, depending on what age is utilized to distinguish between adult and pediatric patients, between 3000 and 4000 children each year are diagnosed. Concepts and management of pediatric brain tumors are constantly evolving [Bleyer, 1999; Jemal et al., 2008; Louis et al., 2007a; Smith et al., 1998]. The classification of these neoplasms, on which treatment is based, is also changing, primarily due to the incorporation of molecular diagnostics.
As in adults, the majority of primary central nervous system tumors in children are glial in origin; however, a significant proportion of tumors are embryonal (Figure 83-1) [CBTRUS, 2008; Jemal et al., 2008]. Embryonal, and mixed neuronal and glial tumors in infants are notoriously difficult to diagnose. The routine utilization of immunohistochemical analysis and, more recently, molecular analysis of tumors have altered concepts of diagnosis and classification, but their incorporation has lagged behind those in other childhood cancers, such as leukemia and neuroblastoma, and has led to the continued subjectivity of diagnosis.
Advances and refinements in therapy, to date, have resulted in a modest improvement in overall survival rate of patients. It has been estimated that there has been a 15–20 percent improvement in survival in patients diagnosed between 1975 and 1979, compared to those diagnosed and treated between 1996 and 2003 [CBTRUS, 2008; Jemal et al., 2008]. The complexities involved in the treatment of childhood brain and spine tumors have resulted in the development of regional centers focusing on the comprehensive management of children with these neoplasms. It is widely accepted that, for optimum management, a multidisciplinary approach is required, involving many specialties, including neurosurgery, neurology, oncology, neuropathology, neuroradiology, radiation oncology, ophthalmology, and rehabilitative specialists. In addition, given the relative infrequency of specific subtypes of childhood brain tumors, for progress to be made expeditiously, prospective multicentered, cooperative group-based trials are optimal. National and, in selected cases, international collaborations have become the cornerstone of modern management of childhood brain tumors.
Incidence
The incidence of childhood nervous system tumors is best estimated from population-based registries. Institution-based registries are potentially flawed by referral patterns; the tendency for larger programs to receive referrals of selective patients, skewing patient number; and the increasing mobility of patients, allowing individuals to seek second opinions and thus being included in statistics at multiple centers. The reported incidence rates of brain tumors for patients between 0 and 19 years ranges from 3.3 to 4.5 cases per 100,000 person years [CBTRUS, 2008]. This reported incidence is higher than the 2.7 cases per 100,000 person years reported in the late 1970s and early 1980s. It is believed that this higher incidence is partially due to the availability of magnetic resonance imaging (MRI), identifying more brain tumors earlier, and possibly the use of stereotactic biopsies to confirm the presence of primary central nervous system tumors [Bleyer, 1999; Smith et al., 1998]. Better reporting, the inclusion of lower-grade tumors in registries, and the wider utilization of epilepsy surgery confirming the diagnosis of low-grade neoplasms may also contribute to this increased incidence, rather than it representing a true increase in the number of brain and spine tumors. However, some studies have suggested that there is indeed a true increase in incidence. Among the major histological groupings, rates are highest for tumors of neuroepithelial tissue, with pilocytic astrocytomas and medulloblastomas being the most common individual subtypes of tumors.
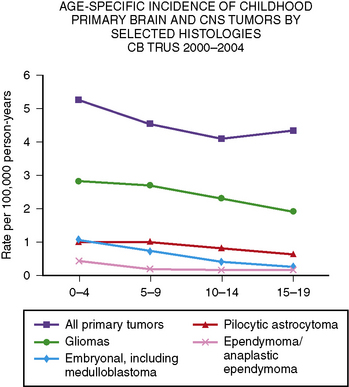
Primary central nervous system tumors are more common in the first decade of life (Figure 83-2) [CBTRUS, 2008]. The reported incidence of brain tumors is higher in whites (4.7 per 100,000 person years) than in blacks (3.0 per 100,000 person years). The overall incidence for all brain tumors is highest among those between 0 and 4 years of age (5.2 per 100,000 person years), and lowest among those between 10 and 14 years of age (4.1 per 100,000 person years) [CBTRUS, 2008]. However, incidence does vary depending on tumor type, with ependymomas and medulloblastomas decreasing with age, and pilocytic astrocytomas peaking at 5–9 years of age. Germ cell tumors increase with age. In the 0–19 years age group, brain tumors are more common in boys, especially tumors of neuroepithelial origin and pineal germ cell tumors.
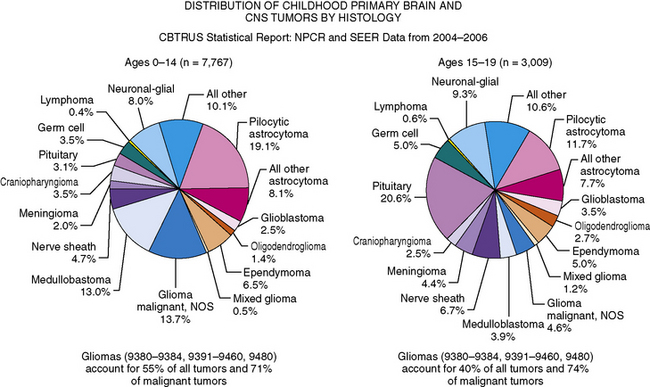
Overall, gliomas of multiple different subtypes, including pilocytic astrocytomas, astrocytomas not otherwise specified, and gliomas, are the most common type of childhood brain tumors, accounting for over 50 percent of all tumors in children between 0 and 14 years of age, and nearly 75 percent of all malignant tumors [CBTRUS, 2008]. Embryonal tumors, including medulloblastoma, comprise 15–20 percent of all tumors in children between 0 and 14 years, occurring in approximately 5 percent of those between 15 and 19 (see Figure 83-2) [CBTRUS, 2008].
Etiology
The causation of the majority of all childhood brain tumors is unknown [Bleyer, 1999; Bunin, 2000]. Despite this, there are genetic conditions and, to a lesser extent, environmental exposures that predispose to the development of tumors [Bleyer, 1999; Bunin, 2000]. Genetic syndromes related to increased development of specific tumor types have provided molecular genetic insights into childhood brain tumors that have been partially translatable to the study of apparently nongenetically inherited tumors. The known syndromes that are associated with the development of childhood brain tumors usually have an autosomal-dominant pattern of inheritance. At times, for unclear reasons, brain tumors will arise in multiple family members [Draper et al., 1977; Fitzgerald, 2000; Kuijten et al., 1993; Malmer et al., 2007; Moschovi et al., 1998].
Neurofibromatosis
Probably the most common genetic syndrome associated with development of primary central nervous system tumors is neurofibromatosis type 1, which is an autosomal-dominant disorder due to mutation of the NF1 gene [Burger et al., 1987; Cawthon et al., 1990; Listernick et al., 1999; Wallace et al., 1990]. The NF1 gene on the long arm of chromosome 17 encodes the neurofibromin protein, which, among other actions, is a tumor suppressor gene [Burger et al., 1987; Listernick et al., 1999; Pollack and Mulvihill, 1997]. Its absence allows ras activation and subsequent downstream intracellular signaling through the RAS/MAPK (MAP kinase) pathway. The gene is large, spanning 350 kb, and a mutation in the initially unaffected (wild-type) NF1 gene is thought to result in the development of a host of tumors. In the central nervous system, the most common are low-grade, most often pilocytic astrocytomas, which early in life have a marked predilection for the optic nerves and other parts of the visual pathway. Later in childhood, gliomas may develop in other central nervous system sites, especially midline areas, including the brainstem and corpus callosum.
Separation of low-grade tumors from areas of brain dysplasia, also common in neurofibromatosis type 1, can be difficult [Listernick et al., 1997]. These areas of abnormal signal on T2-weighted MRI are at least partially developmental, and many disappear as the child enters into puberty. Other central nervous system tumors, such as ependymomas and possibly medulloblastomas, are more common in patients with neurofibromatosis type 1. Neurofibromatosis type 1 occurs in as many as 1 in 3500 patients at risk, and brain tumors may occur in as high as 20 percent of patients, although, for unclear reasons, many affected patients do not require specific treatment.
Children with neurofibromatosis type 1 are also prone to development of neurofibromas, of which the most problematic in prepubertal patients are plexiform neurofibromas [Listernick et al., 1999; Packer and Rosser, 2002; Packer et al., 2002a]. Plexiform neurofibromas are congenital and may cause significant cosmetic disfigurement, organ dysfunction, and, dependent on location, neurologic compromise, including spinal cord compression. A percentage of plexiform neurofibromas, possibly as high as 10 percent, will undergo malignant transformation, although in the vast majority of patients this occurs after puberty, especially in the early adult years. Since patients with neurofibromatosis type 1 have an increased predilection for the development of other malignancies, including myelogenous leukemia and sarcomas, treatment of patients with neurofibromatosis type 1-associated brain tumors can be quite challenging. Chemotherapeutic alkylating drugs and radiation therapy may increase the risk of developing secondary tumors.
Neurofibromatosis type 2, which is also an autosomal-dominant disorder affecting approximately 1 in 25,000 to 1 in 400,000, is due to a gene abnormality on the short arm of chromosome 22 [Pollack and Mulvihill, 1997; Rouleau et al., 1993; Trofatter et al., 1993]. Patients with this condition are predisposed to developing schwannomas of the cranial nerves, spinal nerves, and peripheral nervous system. Bilateral acoustic schwannomas are essentially pathognomonic of the illness and are usually present by early adulthood. Gliomas and ependymomas, especially spinal ependymomas, also occur with increased frequency.
Tuberous Sclerosis and Other Autosomal-Dominant Conditions
Other autosomal-dominant neurogenetic diseases are related to specific tumor types. Tuberous sclerosis is associated with the development of subependymal giant cell astrocytomas, which tend to occur in the subependymal ventricular zone and most commonly cause obstructive hydrocephalus early in illness [Kandt et al., 1992; van Slegtenhorst et al., 1997]. Cerebellar gangliocytomas are associated with Cowden’s syndrome, caused by a germline mutation of the PTEN gene [Liaw et al., 1997]. The occurrence of dysplastic gangliocytoma of the cerebellum is also known as Lhermitte–Duclos disease. Caused by a mutation of the VHL gene, von Hippel–Landau syndrome is associated with the development of hemangioblastomas in the cerebellum, spinal cord, and retina [Duan et al., 1995; Latif et al., 1993].
Li–Fraumeni Syndrome
Li–Fraumeni syndrome is caused by a germline mutation in the multifunctional TP53 gene [Malkin et al., 1990; Srivastava et al., 1990]. The protein encoded by the gene has a role in cell-cycle control and in DNA integrity and repair. Gliomas, both high-grade and low-grade, are most common in patients with Li–Fraumeni syndrome, and such tumors may be multifocal. Other central nervous system tumors that occur in increased incidence in this syndrome include medulloblastomas, supratentorial neuroectodermal tumors, and choroid plexus tumors.
Gorlin’s Syndrome
Gorlin’s syndrome, also known as the nevoid basal cell carcinoma syndrome, accounts for probably less than 1 percent of all patients with medulloblastoma, but the relationship between this condition and the development of medulloblastoma has led to a better understanding of the molecular pathogenesis of a larger subset of patients with medulloblastoma [Amlashi et al., 2003; Stavrou et al., 2001; Taylor et al., 2000]. It has been estimated that 3–5 percent of patients with Gorlin’s syndrome will develop medulloblastoma. Gorlin’s syndrome is caused by an inherited germline mutation of the PATCHED 1 (PTCH1) gene on chromosome 9, which encodes the sonic hedgehog (SHH) receptor. PTCH1 normally suppresses sonic hedgehog signaling [Taylor et al., 2000]. The syndrome may be diagnosed early in life by characteristic dermatological and skeletal features, including odontogenic keratocysts of the jaw, bifid or fused ribs, macrocephaly, and calcification of the falx. The basal cell carcinomas tend to occur later in life. Mutations of the PTCH1 gene can be demonstrated in 20–40 percent of medulloblastomas, especially in the desmoplastic variant in infants. Since patients with Gorlin’s syndrome are predisposed to the development of basal cell carcinomas later in life, the use of craniospinal radiotherapy, which is the backbone of treatment for most patients with medulloblastoma, is problematic, as basal cell carcinomas may occur in large numbers in the radiation fields. The diagnosis of Gorlin’s syndrome is often made later in life, when the basal cell tumors become evident.
Turcot’s Syndrome
Turcot’s syndrome is another autosomal-dominant disorder due to mutations in the adenomatous polyposis coli (APC) gene (type 2), or secondary to mutations in DNA mismatch repair genes HPS2 and MLH1 (type 1) [Attard et al., 2007; Foulkes, 1995; Hamilton et al., 1995; Ng et al., 2005]. Patients with mutation of a mismatch repair gene are prone to developing colorectal carcinomas and gliomas. Patients with type 2 disease are at increased risk for developing medulloblastomas. Mutations in the APC gene result in abnormalities of the Wnt molecular pathway. This pathway has been noted to be abnormal in up to 15 percent of patients over 3 years of age with medulloblastoma, especially older patients, and may constitute a different, more favorable form of the disease.
Environmental Exposures, Including Radiation Therapy
Of the environment exposures that predispose to the development of brain tumors, the best documented is ionizing radiation [Bakhshi et al., 2003; Kimball Dalton et al., 1998; Rimm et al., 1987; Ron et al., 1988]. This association was demonstrated in children treated with radiation for tinea capitis during the 1940s and 1950s, who were found to have an increased incidence, later in life, of meningiomas, gliomas, and nerve sheath tumors [Ron et al., 1988]. Secondary brain tumors have been reported after cranial irradiation for leukemia, head and neck tumors, and primary brain tumors [Kimball Dalton et al., 1998; Kleinerman, 2009; Neglia et al., 1991; Rimm et al., 1987; Thierry-Chef et al., 2008]. The most common form of malignant brain tumor occurring after radiation is malignant gliomas, which tend to appear 5–15 years after radiation and are highly resistant to therapy. Meningiomas are the most common radiation-induced tumors, increase in frequency with time after radiotherapy, and do not begin to peak until ten years after exposure [Neglia et al., 1991; Rimm et al., 1987]. Other tumor types that have been reported include lower-grade gliomas, medulloblastomas, and ependymomas.
The effects of a multitude of environmental exposures, including maternal diet, on the occurrence of childhood brain tumors have not been conclusively shown [Bunin, 2000; Preston-Martin, 1996]. Factors that have been associated with a higher incidence of medulloblastoma in some, but not all, studies include maternal farm residence during pregnancy and pesticide exposure [Connelly and Malkin, 2007; Norman et al., 1996; Preston-Martin, 1996; Robison et al., 1995]. In some studies, mothers’ multivitamin use and food consumption during pregnancy have been found to be potentially protective against of the development of tumors, especially medulloblastoma. Initial studies suggested an association between significant exposure to electromagnetic waves or the use of cell phones in the development of gliomas, but other studies have not been able to confirm this relationship [Ali Kahn et al., 2003; Connelly and Malkin, 2007; Hardell et al., 2002]. Similarly, initial studies suggested that patients exposed to simian virus 40 (SV40), through immunizations, may have a higher incidence of developing medulloblastoma [Hayashi et al., 2001; Kim et al., 2002]. Subsequent studies could not confirm this association. Immunosuppression, either due to an underlying disorder of the immune system or after treatment with chronic immunosuppressive agents in situations such as organ transplantation, has been associated with an increased incidence of central nervous system lymphoma [Kuijten and Bunin, 1993; Penn, 1998].
Pathology and Classification
The present classification of primary central nervous system tumors dates back to the 1920s and, as devised by Bailey and Cushing, was based on a cell of origin concept and the theory that tumor cells developed from cells arrested in various stages of development [Bailey, 1925; Louis et al., 2007b]. For each developmental stage of a cell, a corresponding tumor could be identified. In the Bailey and Cushing classification, tumors were also classified on the basis of the morphology of the tumor [Bailey, 1925]. This classification was modified by histologic grading of predominantly glial tumors on a 1–4 scale, with 1 being the most benign and 4 being the most malignant. Over time, other systems have been devised, essentially to score tumors to connote their relative aggressivity and to link the nomenclature to prognosis.
At present, the most widely accepted classification schema is that of the World Health Organization (WHO) [Louis et al., 2007a]. This system, since its first publication in 1979, has included a grading scheme that basically attempts to be a malignancy scale. Generically, grade 1 tumors are those with low proliferative potential and the possibility of cure following surgical resection alone. Grade 2 lesions are believed to be more infiltrative in nature and may have a higher likelihood of recurring. Dependent on the tumor type, grade 2 tumors are also those considered to more likely to progress to higher grades, especially in the case of gliomas. Grade 3 is generally reserved for tumors with histological evidence of malignancy, which are less likely to be cured by surgery alone. Grade 4 tumors are those that are considered the most cytologically malignant and mitotically active. There have been attempts to modify the WHO classification for use in pediatrics. Although the WHO classification is applicable to pediatrics, the congenital nature of many tumors, often with elements that seem to show mixed lineage, such as neuronal and glial elements, can make classification difficult. In addition, in the pediatric modifications, which have not been fully accepted, tumor location is also a component of classification.
Probably the most controversial aspect of the classification of pediatric brain tumors has been the nomenclature utilized for central nervous system embryonal tumors [Rorke, 1983]. The most common embryonal tumor is the medulloblastoma, a term that was coined over 80 years ago and still exists, although it is now accepted that there is no such tumor cell as a medulloblast. The primary controversy centered on whether all histologically similar, primitive neuroectodermal tumors should be classified as one tumor type and then subclassified on the basis of other histological features, such as histological differentiation along one or more cellular lineages, or whether the tumor should be diagnosed based on both histological features and tumor location. The introduction of immunohistochemical techniques demonstrated the variable cell types that occur in embryonal tumors, although the majority of cells are small hyperchromatic round or oval cells (small blue cells) [Gould et al., 1990; Molenaar et al., 1989]. The availability of molecular genetic analysis helped settle this controversy, as it was shown that primitive embryonal tumors occurring in the cerebellum were molecularly distinct from those arising in the pineal region and from those arising in the cortex [Ellison, 2002; Haberler et al., 2006; Pomeroy and Sturla, 2003; Rorke et al., 1995].
The WHO classification does not separate tumor types on the basis of tumor location [Louis et al., 2007a]. However, by convention, childhood central nervous system gliomas are often discussed by location, and cerebellar astrocytomas, intrinsic brainstem gliomas, and visual pathway gliomas, especially optic nerve and/or chiasmatic tumors, are considered relatively distinct entities. There are few biologic data to support such separations, and there is probably little, if any, prognostic difference between a diffuse intrinsic brainstem glioma and a similar tumor involving the thalamus, or, for that matter, a juvenile pilocytic astrocytoma of the cerebrum versus juvenile pilocytic astrocytoma of the cerebellum.
Medulloblastoma and Supratentorial PNET
Isochromosome 17q, often in combination with loss of 17p, is the most common cytogenetic alteration in medulloblastoma, occurring in up to 50 percent of tumors [Biegel, 1999]. A clear association with clinical outcome has not been confirmed; however, recent evidence suggests that isochromosome 17q is an unfavorable prognostic indicator [Pan et al., 2005]. Supratentorial primitive neuroectodermal tumors (sPNET) display a unique set of genomic alterations that rarely include isochromosome 17q, indicating that sPNET are molecularly distinct from medulloblastoma, despite sharing a similar histological appearance [Bayani et al., 2000; Pfister et al., 2007].
High expression of the neurotrophin-3 receptor, TRKC, was the first molecular alteration identified as an independent predictor of favorable outcome in medulloblastoma [Grotzer et al., 2000; Segal et al., 1994]. Increased expression of ErbB2, a member of the epidermal growth factor receptor (EGFR) family, and MYCC oncogene independently correlates with a poor outcome [Gilbertson et al., 1995; Grotzer et al., 2001]. High-level amplifications of MYC family members are also seen in 5–15 percent of medulloblastomas, and are associated with the aggressive anaplastic medulloblastoma variant and unfavorable outcome [Herms et al., 2000].
Aberrant signaling through the developmental pathways, Hedgehog (Hh), Notch, and Wnt, has been linked to medulloblastoma formation and growth (Figure 83-3). The relationship between Hh and medulloblastoma was deduced from the observation that medulloblastomas arise in patients with nevoid basal cell carcinoma syndrome (Gorlin’s syndrome), a condition caused by a somatic mutation of the PATCHED (PTCH) gene [Wicking and Bale, 1997]. PTCH is an important regulator of the Hh mitogenic signaling pathway. Gorlin’s syndrome accounts for 1–2 percent of all medulloblastomas, most commonly of the desmoplastic subtype [Amlashi et al., 2003]. Mutations in Hh pathway genes have been identified in approximately 25 percent of sporadically occurring medulloblastomas, predominantly of the desmoplastic subtype [Dong et al., 2000; Pomeroy et al., 2002; Raffel et al., 1997].
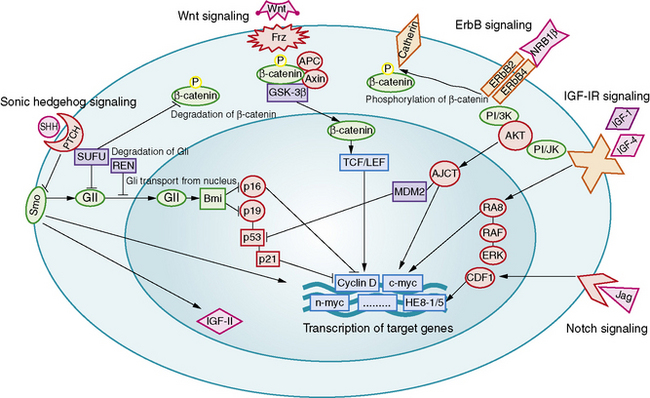
Increased copy numbers of Notch and mutations in the Wnt signaling pathway have each been detected in up to 15 percent of medulloblastomas. Expression of the Notch pathway target gene Hes1 is associated with shorter survival time, while nuclear immunoreactivity for beta-catenin, a marker of Wnt pathway activation, is associated with a highly favorable outcome [Ellison et al., 2005; Fan et al., 2004]. A more recent integrative genomic study identified five distinct molecular subtypes of medulloblastoma, including a Wnt signaling group associated with classic histology tumors in older children, and an Hh signaling group associated with desmoplastic tumors in younger children [Kool et al., 2008]. Both tumor groups shared increased expression of Notch and platelet-derived growth factor (PDGF) pathway genes.
Astrocytoma
Perhaps the most significant discovery to date for this group of tumors is the identification of BRAF oncogene-activating mutations in association with juvenile pilocytic astrocytoma (JPA) [Jones et al., 2008]. BRAF mutations appear to be rare in other types of pediatric low-grade glial tumors, and are associated primarily with JPA of the cerebellum and optic pathway [Jacob et al., 2009]. Low-grade gliomas, usually JPA of the optic nerves and chiasm, also occur in about 15 percent of children with neurofibromatosis type 1; however, BRAF mutations have not been identified in neurofibromatosis type 1-associated JPA [Jacob et al., 2009].
Evidence shows that childhood and adult malignant gliomas are molecularly distinct. Overexpression of EGFR is seen in 60–80 percent of high-grade gliomas in both children and adults; however, amplification of EGFR is found in 40 percent of adult tumors, but only rarely seen in childhood gliomas [Bredel et al., 1999]. TP53 is mutated in 12 percent of malignant gliomas in children aged less than 3 years, compared to 40 percent of the tumors in children older than 3 years [Pollack et al., 2001]. The 5-year survival is 17 percent in patients with high TP53 expression, and 44 percent in those with tumors that have low TP53 expression [Pollack et al., 2002]. PTEN mutations occur in only 8 percent of high-grade lesions in children, compared to 30 percent of adult high-grade tumors; however, expression of activated PDGF receptors and PTEN deficiency are associated with increasing malignant histology and decreased survival, respectively, among children with high-grade gliomas [Cheng et al., 1999; Raffel et al., 1999; Thorarinsdottir et al., 2008].
Other Childhood Brain Tumors
One of the most important molecular genetic alterations in childhood brain tumors is the discovery of hSNF5/INI1 gene mutations in association with atypical teratoid/rhabdoid tumors (AT/RT) [Biegel et al., 2002a]. AT/RT has sometimes been confused with medulloblastoma and PNET, but now screening for hSNF5/INI1 mutations can confirm the molecular diagnosis of AT/RT, which has important prognostic and therapeutic implications.
In ependymoma, chromosome 17p deletions have been found in 50–80 percent of tumors, but TP53 mutations have not been found to date [von Haken et al., 1996]. An integrative genomic study of ependymoma also revealed that supratentorial, posterior fossa, and spinal tumors have distinguishable molecular signatures, and that genes of supratentorial and spinal tumors are similarly expressed in mouse radial glial cells in the corresponding region of the CNS [Taylor et al., 2005].
Neuroradiology
The ability to stage tumor extent – especially to detect the presence of subarachnoid metastatic dissemination – is crucial. Many pediatric brain tumors have a propensity to disseminate throughout the subarachnoid spaces, often at the time of presentation. High-quality screening MRI studies of the entire neural axis are needed; clear outcome differences have been demonstrated with regard to pediatric brain tumors, depending on the presence or absence of dissemination [Packer et al., 2006]. Finally, repeated follow-up at appropriate time intervals is the best method for detecting early tumor recurrence; detection of smaller, presymptomatic recurrent tumor leads to improved outcomes [Saunders et al., 2003; Shaw et al., 1997].
Diffusion Imaging
Diffusion imaging (DI) investigates molecular translational movements (i.e., Brownian motion) of water molecules. Brownian motion produces signal loss proportional to the degree of molecular translation/diffusion. Within the cerebral milieu, cellular membranes and other macromolecular structures restrict water diffusion, which results in increased signal on DI. (Less diffusion means less signal loss secondary to spin dephasing.) Thus, signal on diffusion images can reflect cellular density and microarchitecture. The apparent diffusion coefficient (ADC) is a measure of the ability of tissue to restrict water diffusion. ADC adversely correlates with the grade of tumor and the cellular proliferation Ki-67 labeling index [Higano et al., 2006]. Increase in ADC, reflecting decrease in cellularity, has been observed as tumors respond to treatment.
In the intracellular and extracellular spaces of the brain, macromolecular proteins, intracellular organelles, cell walls, and myelin sheaths restrict or slow diffusion in certain directions. The most significant determinant of the preferred diffusion direction is the myelin sheath within cerebral white matter [Miller et al., 2003]. DI can therefore assess the microscopic structural integrity of white-matter tracts as the movement of water occurs preferentially along the long axis of the myelin sheath surrounding the axon. Anisotropy is an aggregate measure of the magnitude of displacement across all three dimensions; using information gleaned from the size, shape, orientation, and distribution (or spatial pattern) of diffusion ellipsoids within an imaged volume, one can characterize features of water diffusion within a voxel – the diffusion tensor. A voxel by voxel analysis of anisotropy can be performed, resulting in diffusion tensor imaging (DTI). Connectivity maps (i.e., tractography) can also be generated, which demonstrate the long association (axonal) pathways that convey cortical connections as tracklike structures.
DTI tractography is used to plan the surgical approach to a focal tumor. Important association pathways (e.g., pyramidal, optic tracts) can be localized in relation to a tumor; this helps the surgeon to avoid injury to these white-matter tracts. DTI also shows promise as a tool to evaluate for the presence and extent of tumoral invasion; whereas focal (well-marginated) tumors should displace but not disrupt axons outside of their borders, the microscopic tumor extension of infiltrative tumors is expected to disrupt local axonal structures [Helton et al., 2006; Talos et al., 2007].
Magnetic Resonance Spectroscopy
Spectroscopy can be performed with single-voxel or multivoxel techniques. Tissue composition within a voxel must be homogeneous, otherwise partial volume averaging of tissue of various compositions (e.g., normal, tumor, and necrotic tissue) occurs and the spectrum has little value. Single-voxel studies have improved spectral resolution over multivoxel techniques, though minimal voxel size for good signal to noise is about 4 cm3; further, in single-voxel imaging, the prescription (i.e., the determination of the voxel location) of the voxel is totally operator-dependent – and therefore not very reproducible. Spectroscopic analysis of a large volume of cerebral tissue is feasible with multivoxel techniques (i.e., two- or three-dimensional chemical shift imaging [CSI]), in which several subcentimeter voxels can be examined simultaneously. CSI is very helpful in the evaluation of small or heterogeneous tumors; in the evaluation of the extent infiltrative tumors where the tumor/normal brain boundary may be difficult to determine with conventional techniques; and to distinguish recurrent tumor from radiation necrosis. MR spectroscopy may be of help to monitor tumor response versus progression [Tzika et al., 2004]; prognosticate overall survival [Warren et al., 2000]; and improve preoperative identification of tumor type [Panigrahy et al., 2006].
Perfusion Imaging
Perfusion imaging can be performed with techniques based on dynamic susceptibility contrast (DSC) or on vascular permeability. DSC techniques assess cerebral tissue perfusion following a dynamic injection of gadolinium. During the first-pass transit of contrast through cerebral capillaries, which lasts anywhere from 5 to 15 seconds, gadolinium is (normally) restricted to the intravascular space. This intravascular restriction gives rise to signal inhomogeneity within image voxels (i.e., a susceptibility effect), which is reflected as signal loss on susceptibility-sensitive T2 (T2*) imaging techniques. A curve showing concentration of gadolinium over time can be generated. This enables a time versus signal intensity curve to be drawn, from which a variety of parameters can be inferred. These include mean transit time (MTT); time-to-peak; area under the curve, a measure of relative cerebral blood volume (rCBV); and relative cerebral blood flow (rCBF). rCBV corresponds to the volume of blood within brain tissue and can reflect the neovascularization associated with tumor growth (tumor angiogenesis). Perfusion imaging is therefore useful for the study of neovascularization and angiogenesis inhibition; for evaluation of tumor grade [Schmainda et al., 2004]; for targeting a lesion for biopsy; and to differentiate hypervascular tumors from tissue necrosis. DSC perfusion imaging may also be able to identify low-grade tumors that progress rapidly, and low-grade gliomas that have a propensity for malignant transformation [Law et al., 2006].
DSC perfusion studies do not consistently generate usable data. First, a larger-bore intravenous catheter must be placed for safe delivery of the injection of 3–4 cc/sec of gadolinium necessary to generate a well-defined first-pass curve; that is not always easy, especially in children with poor intravenous access. (Recent commercial introduction of ports and central lines that are safe to use with power injectors may alleviate this issue.) Second, susceptibility artifacts are not negligible, as DSC perfusion utilizes an echo planar technique (echo planar images are prone to image distortion by susceptibility artifacts); studies performed on children with dental braces or in the presence of significant amounts of intracranial/intratumoral hemorrhage are often useless. Finally, gadolinium contrast can leak through tumor capillaries; such leakage can result in an underestimation of rCBV, as the computations performed to process the susceptibility T2* signal assume that the contrast remains confined to the capillary [Cha et al., 2002].
Permeability-based perfusion techniques take advantage of leakage of gadolinium through tumor vessel walls. In healthy cerebral capillaries, the blood–brain barrier prevents gadolinium leakage into the interstitium. Tumor capillaries have disrupted or absent blood–brain barriers, which allows contrast to leak outside the vessel. The rate of leakage is proportional to the permeability of the capillary wall. Permeability imaging, also known as relaxivity imaging, is a T1-weighted technique in which a curve of dynamic signal intensity (i.e., enhancement) versus time is generated. The slope of enhancement reflects the degree of permeability of capillaries. The intensity of initial enhancement reflects tissue vascularity; fractional blood volume (fBV) can be extrapolated [Roberts et al., 2000]. fBV is a fairly good estimate of the rCBV data obtained with susceptibility perfusion [Haroon et al., 2007].
Positron Emission Tomography
Positron emission tomography (PET), with use of (18)F-fluorodeoxyglucose (FDG), allows for the evaluation of regional cerebral glucose metabolism. PET is useful in the determination of tumor metabolism/grade and of tumor extent, and to distinguish persistent or recurrent tumor from radiation necrosis [Pirotte et al., 2007]. Metabolic changes can be measured with a spatial resolution approaching 2–3 mm in the newer scanners. Images can be co-acquired with a CT scan (CT-PET), or can be co-registered with MR images using fusion software. Metabolic activity of tumor sites can be determined with quantitative, as well as qualitative (comparing FDG uptake in a tumor to uptake of normal gray or white matter), assessment. Higher-grade tumors tend to be more glucose-avid than lower-grade tumors, except for juvenile pilocytic astrocytomas, which show very high glucose uptake. For lesions that do not show abnormal glucose uptake, of (11)C-methionine PET scanning is helpful to differentiate neoplastic from non-neoplastic types. Thallium-201 single-photon emission CT (SPECT) now has a decreasing role in evaluation of pediatric brain tumors, given its more limited anatomic resolution.
Staging and Stratification
Pre- and postoperative staging is the cornerstone of therapy for most embryonal childhood central nervous system tumors and, to a lesser extent, glial neoplasms [Gajjar et al., 2004; Packer et al., 1999a]. The utility of staging for both determination of prognosis and decisions concerning most appropriate therapy has been best shown for medulloblastoma, as staging has been an accepted component of management since the mid-1980s. The first widely accepted staging system was the “Chang” classification schema, which has been extensively modified over time. All these schemas share the property of being based on degree of surgical resection or, more specifically, the amount of tumor left after surgery and the extent of tumor spread outside the primary tumor site. The latter is determined by a combination of results from MRI of the entire brain and spine, and cerebrospinal fluid cytological examination, preferably from the lumbar space, if deemed medically safe [Deutsch and Reigel, 1980; Gajjar et al., 2004; Packer, 1999; Packer et al., 1999a]. Embryonal tumors and glial neoplasms rarely spread to non-neural sites; thus, evaluation for disease outside the primary nervous system, at the time of diagnosis, is usually not indicated. The classical TNM (tumor, nodes, metastasis) staging system used for non-central nervous system tumors is replaced by a TM system for brain and spine tumors. Although the need for lumbar cerebrospinal fluid cytological examination has been debated, studies have shown that it provides complementary information to imaging results [Packer et al., 1999a; Zeltzer et al., 1999].
A complicating factor has been the fact that the performance and interpretation of imaging results have been variable in national and international studies [Packer et al., 2006]. In the most recent medulloblastoma studies, approximately 10 percent of studies were inadequately performed, primarily due to movement artifact or incomplete imaging (primarily of the spine), and another 5–10 percent of studies are considered, upon central review, to be incorrectly interpreted. Tumors that are nonenhancing at the primary site may result in nonenhancing dissemination along the neuroaxis, which is difficult to assess and leads to under-assessment of the extent of disease. In addition, normal superficial spinal cord vascularity can be misinterpreted as leptomeningeal enhancement.
Histology has been incorporated into risk stratification for many tumor types. For embryonal tumors, such as medulloblastoma, histological features, including desmoplasia/nodularity in infants and anaplasia in infants and older children, are now increasingly accepted as important components of staging and risk-stratification classifications, although the independent significance of such features has been related variably to outcome [Eberhart et al., 2004; Giangaspero et al., 1999, 2006; Grotzer et al., 2007]. For glial tumors or ependymomas, histological features that have been translated into grades are an accepted component of classification and are used both for risk stratification and for determinations of treatment.
It has become clear for the embryonal tumors, especially medulloblastoma, that molecular genetic parameters provide extremely important information [Ellison et al., 2005; Fernandez-Teijeiro et al., 2004; Gajjar et al., 2004; Grotzer et al., 2007; Pfister et al., 2009; Ray et al., 2004]. In retrospective studies, a host of chromosomal and molecular genetic findings had been related to outcome. In some cases, these findings show a strong correlation with other parameters, such as the tendency to disseminate or features of anaplasia, but in other studies they are of independent prognostic value.
Clinical Presentation
Nonlocalizing Signs and Symptoms
Nonlocalizing signs and symptoms predominate early in the course of illness in almost all types of childhood nervous system tumors. This is due to a variety of factors, including the predilection of childhood brain tumors to arise in the posterior fossa and cause obstruction of cerebrospinal fluid early in the course of illness, resulting in the nonspecific symptoms of headaches, nausea, vomiting, and lethargy. Separation of headaches caused by neoplasms, as opposed to the more common headaches children develop, can be extremely difficult [Glaser et al., 1971; Honig and Charney, 1982]. The majority of children with brain tumors, especially those with posterior fossa tumors and large supratentorial tumors, will have headaches by the time of diagnosis. However, headaches are notoriously nonspecific early in the course of illness. A change from a nonspecific headache pattern to morning headaches should raise concern that a mass lesion is present. Headaches that wake a child from sleep also suggest the possibility of increased intracranial pressure. Although this is true in general terms, recognizing such changes, in practicality, can be quite difficult for the child or family, and migraine headaches can wake a child from sleep. Headaches are especially difficult to characterize in very young children.
Localizing Symptoms
Supratentorial Tumors
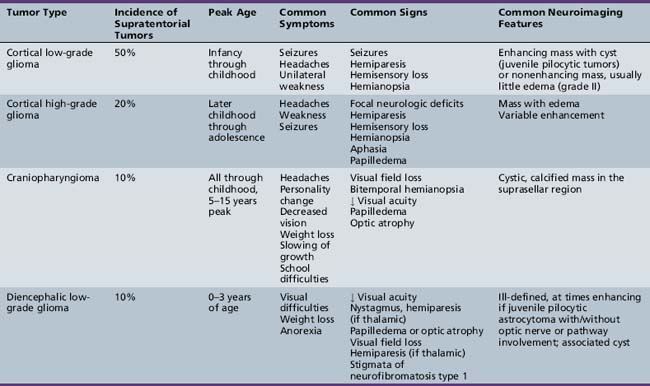
The diagnosis of primary central nervous system tumors is usually easier when there are associated localizing signs and symptoms (Table 83-1). Supratentorial tumors present with symptoms and signs similar to those seen in older patients and adults. Headaches are the most common initial complaint and are often nonlocalizing early in the course of illness [Hirsch et al., 1989]. Seizures are the second most common symptom encountered and approximately one-quarter of children with supratentorial tumors will have seizures as their initial symptom. This is especially true with tumors of the frontal or temporal lesion. Slower-growing cortical tumors, especially gliomas, are more likely to result in convulsions early in the course of illness. Although patients with more aggressive cortical gliomas may have seizures, focal neurologic deficits, such as hemiparesis or, in a patient amenable to careful examination, hemisensory loss or visual field loss, are more common.
Posterior Fossa Tumors
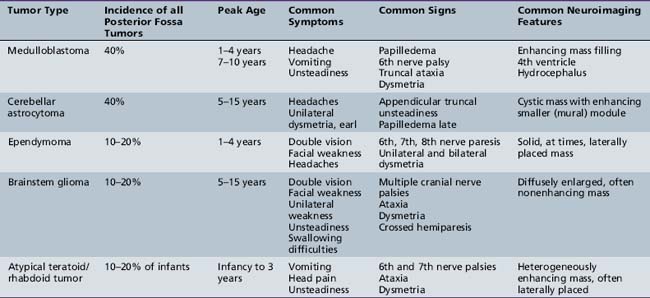
Five tumor types are most likely to occur in the posterior fossa, as noted in Table 83-2. Since cerebellar astrocytomas are more laterally placed and are slower-growing tumors, their symptoms tend to be more indolent than those of medulloblastomas, which fill the fourth ventricle early in the course of illness and result in obstructive hydrocephalus. However, midline cerebellar astrocytomas may mimic medulloblastomas [Packer et al., 1999a]. Brainstem gliomas are an insidious lesion and are likely to cause cranial nerve neuropathies associated with motor, cerebellar, and sensory deficits. Ependymomas are the great mimickers of other tumor types in the posterior fossa and may present as slow-growing, medulloblastoma-like tumors or, if the tumor involves the cerebellopontine angle, can mimic brainstem gliomas and result in multiple cranial neuropathies, especially sixth, seventh, and eighth nerve palsies.
Midline Tumors
The infiltrating nature of midline tumors may also make diagnosis difficult [Albright et al., 1984; Glaser et al., 1971]. Many of these neoplasms are low-grade and classically result in insidious, nonspecific, and often nonlocalizing symptoms early in the course of illness. In the school-age child, infiltrating tumors and occasionally midline compressive masses, such as craniopharyngiomas, may also result in the nonspecific finding of declining academic performance, associated with fatigue and personality change.
General Aspects of Treatment
Surgery
Surgery remains the initial step in the treatment for the overwhelming majority of childhood central nervous system tumors, and the degree of resection, even for malignant neoplasms that require other forms of adjuvant therapy, is often an important factor in determining outcome [Albright et al., 1995; Campbell et al., 1996; Chen et al., 2007; Horn et al., 1999; Packer et al., 1999a; Roujeau et al., 2007]. As regards diagnosis, attainment of a representative portion of the neoplasm for histological and increasingly biological analysis is crucial. Exceptions to the need for tissue for diagnosis include neuroradiographic characteristic diffuse intrinsic brainstem tumors; chiasmatic/hypothalamic infiltrating gliomas in children with neurofibromatosis type 1 and possibly for those without; and germ cell tumors, predominantly mixed germ cell tumors, which secrete diagnostic proteins into cerebrospinal fluid and blood.
For some tumor types, surgery is curative and complete resection obviates the need for alternative therapies [Schneider et al., 1992]. This is primarily true for low-grade gliomas, especially pilocytic astrocytomas of the cerebellum or cerebrum, gangliogliomas, other mixed neuronal-glial tumors, and possibly supratentorial low-grade ependymomas [Poussaint and Rodriguez, 2006]. For other, more aggressive tumor types, surgery may be a crucial component of multimodality treatment, as outcome is statistically better for patients with nondisseminated medulloblastomas, posterior fossa ependymomas, and cortical malignant gliomas if total or near-total resection can be obtained [Albright et al., 1989; Packer et al., 1992; Wisoff et al., 1998]. For those tumors that located in eloquent areas of brain or are disseminated, the preoperative assessment of whether “total” resection or an attempt at total resection provides a survival benefit is usually the prime determinant of whether aggressive tumor removal is tried or less aggressive approaches, including open or stereotactic needle biopsy, will suffice. Stereotactic biopsy techniques, be they CT- or MRI-guided, are highly accurate in targeting deep-seated lesions and are associated with morbidities of less than 5 percent and mortalities of less than 1 percent [Broggi et al., 1983]. The advent of biologic therapies, often requiring fresh frozen tissue for molecular analysis, has resulted in the need, in certain circumstances, for additional tissue that cannot be obtained by needle biopsy alone.
Intraoperative Techniques
For those tumors in which extent of resection is a major determinant of outcome, improvements in neurosurgical technology have made “total resections” more likely and possibly safer. Progressive improvements in the operative microscope, which facilitates the visualization of the interface between the neoplasm and normal brain, have allowed for more extensive resections. Frame-based and frameless stereotactic guidance systems are now routinely used for improved preoperative targeting of the tumor and to decrease injury to the surrounding normal brain [Barnett et al., 1993; Kelly, 1990]. Similarly, ultrasonic guidance has allowed for better intraoperative localization. A recent addition to the neurosurgical armamentarium has been the availability of intraoperative MRI, which allows real-time evaluation for extent of surgical resection [Martin et al., 2000]. Intraoperative MRI is performed both with low-field units, which require interruption of the operation for set-up and visualization, and with high-field units, which utilize MRI-compatible surgical equipment and allow the operation to be carried out essentially within the MRI unit. These techniques are most useful for circumscribed benign tumors; however, for infiltrating neoplasms the extent of resection of the neoplasm continues to be difficult to determine.
In functionally critical areas of the brain, intraoperative monitoring of the visual, auditory, and somatosensory pathways can be used to improve the safety of tumor resection [Schneider et al., 1993]. Similarly, for intrinsic and extrinsic brainstem lesions, intraoperative monitoring of cranial nerve function is an important adjunct [Berger et al., 1990]. Maintenance of function intraoperatively does not assure normal function postoperatively, as edema and apparent delayed damage may occur. Awake craniotomy can be used in older children, but it is usually of limited utility in the majority of pediatric brain tumor cases.
Perioperative Considerations
It has been suggested by many that improvements in preoperative and postoperative care have resulted in lower surgical morbidity and mortality. However, what constitutes acceptable morbidity is extremely difficult to define, and obviously, one of the goals of surgery is to have no mortality and limited morbidity. Preoperatively, in children with increased intracranial pressure, every attempt is made to try to decrease pressure and stabilize the patient. In patients with hydrocephalus, this often includes the placement of a temporary external ventricular drain [Dias and Albright, 1989]. In most centers, in patients with posterior fossa tumors and hydrocephalus, a temporary drain is utilized and, within 24–72 hours, the tumor is removed. Removal may restore adequate cerebrospinal fluid flow to avoid and the need for permanent ventriculoperitoneal diversion (ventriculoperitoneal shunting). The minority of patients with posterior fossa lesions, usually fewer than 40 percent, require permanent external drainage. Third ventriculostomy has been proven to be a useful adjunct in reducing the need for permanent ventriculoperitoneal drainage, but the effect of this technique on long-term outcome has not yet been shown [Hopf et al., 1999; Jones et al., 1990].
In children with cortical tumors, prophylactic anticonvulsants are often utilized, even if there is no history of seizures. The need for such treatment and the duration of treatment, postoperatively, are unclear [Foy et al., 1992]. Phenytoin is often used preoperatively due to its lack of significant sedation in most patients, its ability to be given and loaded intravenously, and the ease of switching from intravenous to oral dosing. In patients on steroids, due to drug metabolism interactions, maintenance of therapeutic levels of phenytoin may be difficult, especially when patients are switched to oral dosing. In children who may require postoperative chemotherapy, levetiracetam is an effective alternative, since it also can be used orally or intravenously, and has the added advantage that it does not undergo hepatic metabolism and, thus, does not interact with the metabolism of most antineoplastic chemotherapeutic agents.
Postoperative Morbidity
Surgery in the suprasellar area is frequently associated with increased postoperative visual impairments, which may include decreases in visual acuity and increased deficits in visual fields [Sanford, 1994]. Hypothalamic abnormalities and significant hormonal deficits secondary to disruption of the hypothalamic–pituitary axis also frequently occur. In large tumors that extend to the subfrontal area, removal of the tumor may cause additional damage and both early and late emotional/personality changes. Hypothalamic injury may also result in abnormalities in satiety control and excessive weight gain, primarily in children with craniopharyngiomas.
Surgery for posterior fossa tumors may result in direct brainstem damage, with resultant cranial nerve deficits and damage to the cerebellum or cerebellar peduncles, with increased ataxia and dysmetria. Such direct brainstem damage may be difficult to separate from the increasingly recognized posterior fossa mutism syndrome, which is a constellation of the delayed onset of mutism (characteristically, a few hours to a day after surgery), hypotonia, cerebellar deficits, supranuclear palsies, emotional lability, and severe irritability [Doxey et al., 1999; Packer et al., 1992; Pollack, 1997]. The exact pathogenesis of the posterior fossa mutism syndrome is unknown. Seen primarily, but not exclusively, after medulloblastoma resection, it is thought to be secondary to unilateral or bilateral cerebellar dentate nuclei damage and/or disruption of critical pathways between the cerebellum and the cortex, especially the premotor and supplementary motor cortices. This syndrome results in permanent sequelae in approximately 50 percent of initially symptomatic patients, including persistent abnormalities in tone, coordination, speech, and cognitive abilities [Robertson et al., 2006; Wells et al., 2008]. The presence of lower motor neuron cranial nerve deficits or MRI features of immediate brainstem injury or cerebellar damage makes direct brainstem injury more likely. Postoperative brainstem edema has been reported to be more common in patients with medulloblastoma who develop posterior fossa mutism syndrome compared to patients who do not.
Radiation Therapy
Radiation therapy is an integral component of treatment for many forms of childhood brain tumors. For children with benign tumors, its use is usually limited to those tumors that are not amenable to gross total resections. For children with more aggressive or malignant tumors, radiation therapy has been the backbone of most curative and palliative therapies, even in those patients whose tumors are totally resected. For medulloblastoma, other embryonal tumors, and germ cell tumors, delivery of radiotherapy to the entire neuroaxis (craniospinal radiotherapy), supplemented with boost radiotherapy to the primary tumor site, is routinely employed to treat neuroaxis sanctuary disease and prevent exoprimary disease relapse, even in patients without evidence of tumor dissemination [Packer et al., 1999b]. Craniospinal radiotherapy has been associated with improved survival, but also significant long-term endocrinologic, growth, and cognitive sequelae. In very young children, especially infants, radiotherapy may result in even more severe sequelae and attempts have been made to delay, if not obviate, the need for radiotherapy by the use of chemotherapy. Chemotherapy is often utilized with radiotherapy in older patients, not only to increase survival, but also to decrease the dose or volume of radiation therapy required for disease control or cure.
Changing Techniques/Delivery
As in other forms of medicine, radiation therapy techniques continue to evolve. Focal and craniospinal radiation treatment planning is increasingly dependent on sophisticated three-dimensional computerized analysis [Ciernik et al., 2003; Hug et al., 2002; Kirsch and Tarbell, 2004; Thornton et al., 1991]. “Conformal” radiation therapy is the standard of care and such three-dimensional treatment planning involves multiple individual fields delivered in a variety of different orientations. Intensity modulated radiation therapy (IMRT) is a form of conformal radiation that, depending on the situation, may provide some advantages over other conformal planning techniques [Leibel et al., 1994]. Stereotactic radiosurgery is a further refinement in the delivery of radiation therapy and can be multifractionated, but it is frequently given in a single fraction, termed “radiosurgery.” Radiosurgery systems that deliver hypofractionated radiotherapy, such as the gamma knife, cyber knife, and modified linear accelerated units (LINAC), are designed to deliver a high radiation dose to a small intracranial target in few fractions (often one fraction) by focusing multiple small radiation beams on the target [Boethius et al., 2002; Chiou et al., 2001; Hadjipanayis et al., 2002]. Radiosurgery is usually utilized to treat smaller tumors, normally 3 cm or smaller, but overlapping fields can be used in somewhat larger tumors. Its use in pediatrics has been relatively limited, but it has been employed with good short-term control for small intrinsic tumors, such as low-grade (e.g., pilocytic) gliomas, and other benign tumors, such as craniopharyngiomas. Radiosurgery results in a greater likelihood of acute and subacute reactions, and has less utility in regions of brain where critical areas could be injured by acute necrosis, or in infiltrating tumors.
Radiation therapy is conventionally delivered by photon particles. Other types of particle beam irradiation have been used in selected circumstances. Electron particles, which have a lesser depth of penetrance, have been utilized for delivery of the spinal portion of craniospinal radiation [Pai Panandiker et al., 2007]. Such beams have the disadvantage of not being able to penetrate as deeply as photon irradiation, but have the advantage of having less radiation scatter. Proton beam irradiation is more widely used. It can be modulated to release its energy at a specific depth, unlike photon beams that deliver dose as they enter and exit the target [Hug et al., 2002; MacDonald et al., 2008; Merchant et al., 2008; Plowman et al., 1999]. Dosimetry studies demonstrate that the surrounding areas receive less irradiation after proton treatment compared to photons. For patients undergoing craniospinal radiotherapy, this decrease in “scatter” can spare radiation to tissues surrounding neuroaxis, such as the thyroid gland, colon, and heart. There are also advantages in some posterior fossa tumors, with sparing of dose to the cochlea, especially in treatment of children with posterior fossa ependymomas. Proton beam units were initially almost prohibitively expensive and few units were available to treat children. With new technologies, there will be greater availability in the near future.
In addition to hypofractionated delivery techniques, such as the gamma knife, there has been interest and experience in using a smaller dose of radiotherapy multiple times during the day. Such “hyperfractionated” radiotherapy is based on the concepts that tumor cells are more likely to enter growth phases when they are radiosensitive than non-tumor cells, and that normal cells are better able to repair radiation-induced damage than tumor cells [Freeman, 1996; Mandell et al., 1999; Packer et al., 1993; Thames et al., 1983]. Radiation delivered more frequently allows the normal cells, but not the tumor cells, to undergo cellular repair and, thus, higher doses of radiotherapy can be “safely” utilized, with less toxicity and more efficacy. Early experience with this approach, using dose fractions ranging from 100–120 cGy, twice daily, to total doses as high as 7800 cGy in pediatrics, were disappointing in children with brainstem gliomas [Packer et al., 1993]. There was no improvement in survival, but an apparently higher incidence of acute sequelae, due to brainstem necrosis. Studies using hyperfractionated radiotherapy, especially in delivery of craniospinal radiotherapy, are on-going, with completed studies showing no clear-cut increased acute toxicity and, possibly, improved disease control.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


