and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Vasculitis of the peripheral nerve (VPN) can occur as an isolated process or more commonly as the manifestation, initial or late, of a systemic disease (recently reviewed Vrancken and Said 2013; Collins et al. 2013). Inflammation affects endoneurial and epineurial microvessels, arterioles, and venules; thrombosis and necrosis are often present resulting in ischemia of nerve with varying degrees of axonal degeneration. The vasculitic processes detected in routine nerve biopsy specimens (sural, peroneal, median) occur in medium- and small-sized vessels as the diameter of these structures range from 10 to 350 μm.
With some exceptions the vasculitides found in nerve biopsies (Vital et al. 2006) in many diseases and syndromes are histologically similar; distinction is usually dependent on clinical criteria or biopsy at other sites. In classifying peripheral nerve vasculitis, it is helpful to separate the infectious etiologies and subdivide the remainder into those diseases in which inflammation selectively affects vessels in nerve and those in which vascular involvement is part of a systemic process, acknowledging that sometimes this separation is difficult to establish. Polyarteritis nodosa (PAN), ANCA-related microscopic polyangiitis, Churg–Strauss angiitis (CSA), Wegener granulomatosis (WG), and rheumatoid vasculitis account for about 50 % of cases with systemic vasculitis neuropathy. Attempts to classify vasculopathic diseases, on the basis of the size and type of vessels involved or through putative pathogenic mechanisms, have been unsatisfying (Collins et al. 2010a). Several schemes are in use and we will draw, for the discussion in this chapter, from the nomenclature of the Chapel Hill Consensus Conference (Jennette et al. 2013) and from the classifications proposed by Vrancken and Said (2013) and Gwathmey et al. (2014). We will review the most important types of peripheral nerve vasculitis, followed by a general discussion of the pathological spectrum of peripheral nerve vasculitis and the differential diagnosis. Table 13.1 classifies vasculitis into isolated vasculitis of peripheral nerves, primary systemic vasculitis, vasculitis of connective tissue disease, and less common vasculopathies.
Table 13.1
Peripheral neuropathy due to vasculitis
Isolated PNS vasculitis |
Primary systemic vasculitis |
Polyarteritis nodosa (PAN) |
Churg–Strauss syndrome |
Microscopic polyangiitis |
Wegener granulomatosis |
Essential mixed cryoglobulinemia |
Behçet’s disease |
Henoch–Schönlein purpura |
Collagen vascular diseases |
Rheumatoid arthritis |
Systemic lupus erythematosus (SLE) |
Sjögren syndrome |
Scleroderma |
Other vasculitis and vasculopathies |
Giant cell arteritis |
Paraneoplastica (hematological or solid malignancy) |
Hypersensitivity vasculitis |
Essential mixed cryoglobulinemia |
Sarcoidosisa |
Lymphomatoid granulomatosisa |
Cholesterol embolus syndromea (Bendixen et al. 1992) |
Infection-associated vasculitis |
Leprosy in ENL reaction |
HIV infection |
CMV related |
Unknown basis |
Lyme disease |
Various arthropod stingsa |
Tuberculosisa (Stubgen 1992) |
Other |
Eosinophilia–myalgia syndromea |
Toxic oil syndromea |
13.1 Clinical Manifestations
13.1.1 Mononeuritis Multiplex
Mononeuritis multiplex is regarded as the classic manifestation of vasculitic neuropathy, as the multifocal process affects nerves throughout the body (more often peroneal, ulnar, tibial, and the sural) including the cranial nerves. Patients experience an abrupt onset of pain, paresthesia, and paralysis in the distribution of a single nerve trunk evolving over hours to days, with newly involved nerves appearing over days to months. Atypical forms of presentation include pure sensory ataxia and radiculoplexopathy (Vrancken and Said 2013). When the lesions are disseminated, and their territories overlap, the presentation is in the form of an asymmetric polyneuropathy. Notwithstanding that biopsy is more likely to be performed in patients with mononeuritis multiplex, an acutely, subacutely, or chronically progressive symmetric distal sensorimotor polyneuropathy, where vasculitis is often not suspected, has been described in 19–76 % of biopsy-proven vasculitic neuropathies (Harati and Niakan 1986; Dyck et al. 1987; Hawke et al. 1991; Wees et al. 1981; Kissel et al. 1985; Panegyres et al. 1990; Said et al. 1988). This indicates that progressive symmetric distal polyneuropathy is a common clinical presentation of vasculitic neuropathy. Experience with vasculitic neuropathy at St. Michael’s Hospital includes 39 cases, for which adequate clinical information was available in 32. Fourteen (44 %) presented with a distal symmetrical pattern, 12 (38 %) with obvious mononeuritis multiplex, 5 (16 %) with asymmetrical polyneuropathy, and 1 was asymptomatic. Evolution can occur rapidly enough to result in an initial clinical diagnosis of GBS (Suggs et al. 1992).
Nerve conductions give evidence of axonal abnormalities and are invaluable for demonstrating the asymmetry and multifocality that are most suggestive of a vasculitic neuropathy (Hawke et al. 1991; Kissel et al. 1985; Olney 1992). Although demyelination is generally not a significant element of the histologic picture, conduction block has occasionally been documented as part of an ischemic or vasculitic neuropathy (Hughes et al. 1982; Jamieson et al. 1991; Kaku et al. 1993). Significant demyelinating electrophysiological features were observed in 3 of 32 adequately documented cases of vasculitic neuropathy identified in our laboratory.
In patients known to have a systemic vasculitic illness, nerve biopsy is of questionable value. Of greater interest to the pathologist are those situations where a diagnosis of vasculitis is unsuspected, either because the disease seems confined to the peripheral nervous system, or because it is not yet fully evident
13.1.2 Nonsystemic (Isolated) Peripheral Nervous System Vasculitis (NSVN)
Vasculitis involving the peripheral nerves can be seen in isolation and results in a peripheral nerve syndrome indistinguishable from that of vasculitic neuropathy in multisystem disease. Approximately 25 % of cases with vasculitis neuropathy are diagnosed with primary vasculitis neuropathy. These patients usually present with a subacute or chronic neuropathy, often with a mononeuritis multiplex, and have few or no systemic abnormalities on history, physical, or laboratory investigation. Cases fulfilling these criteria are found in all large series of vasculitic neuropathy (Dyck et al. 1987; Hawke et al. 1991; Vincent et al. 1985; Harati and Niakan 1986; Kissel et al. 1985; Panegyres et al. 1990), and the pathogenesis is probably heterogeneous, as the pathology seems to be (vide infra). It has been argued that these cases do not represent a truly isolated PNS disease because a very high incidence of concurrent vasculitis was found in muscle biopsy (Said et al. 1988). The apparent selectivity of peripheral nerve involvement may reflect an increased vulnerability of this tissue to multifocal microvascular insults, and the presence of a milder disease sufficient to clinically affect nerve but no other tissues might explain the better outcome observed in these patients (Said et al. 1988; Said 1989; Dyck et al. 1987). Some authors have suggested that NSVN should be considered a low-grade systemic vasculitis that is symptomatic in nerves only (Said and Lacroix 2005).
13.1.3 Primary Systemic Vasculitis
13.1.3.1 Classic Polyarteritis Nodosa (PAN)
Classic polyarteritis nodosa (PAN) is a necrotizing arteritis associated with fibrinoid change. It is not uncommonly seen in patients suffering from hepatitis B infection (70 % of cases) and, less often, in patients with hepatitis C or HIV infections (Siva 2001). Although in its presentation PAN may appear limited to skin, muscles or nerves, PAN is a primary systemic vasculitis. Mononeuritis multiplex is recognized in 50–67 % of patients, and it may be the presenting manifestation in most (Hawke et al. 1991; Guillevin et al. 1988; Chumbley et al. 1977; Frohnert and Sheps 1967; Vrancken and Said 2013). Positivity for pANCA is exclusionary for classic PAN. Involvement of medium-sized vessels in epineurium is the domain of classic PAN.
13.1.3.2 Churg–Strauss Angiitis (CSA)
Churg–Strauss angiitis (CSA) is also designated eosinophilic granulomatosis with polyangiitis. In a large series of VPN, CSA accounted for the largest number of cases followed by PAN and WG (Mathew et al. 2007). This disease affects small- and medium-sized vessels. Patients display prominent pulmonary symptoms and eosinophilia, and vasculitic neuropathy develops in 20–65 % of cases, which can be the initial manifestation (Hattori et al. 2002; Uchiyama et al. 2012; Vrancken and Said 2013), CSA is a member of the family of ANCA-associated vasculitides that also includes WG and MPA. Perinuclear ANCA/MPO-ANCA is the pattern most often detected in CSA. The necrotizing vasculitis exhibits PAN-like fibrinoid change and an eosinophilic-rich lymphocytic and histiocytic infiltrate. Extravascular necrotizing granulomata are a feature of CSA, but not PAN (Chumbley et al. 1977).
13.1.3.3 Wegener Granulomatosis (WG)
Wegener granulomatosis (WG) (also termed granulomatosis with polyangiitis) is not a primary vasculitis, but rather a systemic disease characterized by necrotizing granulomata and granulomatous cytoplasmic-ANCA/PR3-ANCA-associated vasculitis (Gross and Csernok 2008; Suppiah et al. 2011) of small- and medium-sized vessels. Paranasal sinuses, lungs, and kidneys (resulting in necrotizing glomerulonephritis) are most severely involved. Peripheral neuropathy, most often with a mononeuritis multiplex pattern due to vasculitis, is the most common neurological manifestation, seen in 10–22 % of patients (Fauci et al. 1983; Drachman 1963; Nishino et al. 1993; Mahr 2009).
13.1.3.4 ANCA-Associated Microscopic Polyangiitis (MPA)
ANCA-associated microscopic polyangiitis (MPA) (formerly designated as microscopic periarteritis nodosa, Wohlwill 1923) is a necrotizing vasculitis affecting predominantly small-sized vessels, associated with antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase (MPO-ANCA) with a perinuclear pattern and sparse immune deposits. The most characteristic clinical feature is rapidly progressive glomerulonephritis, pulmonary involvement, and palpable purpura due to cutaneous vasculitis. Peripheral neuropathy (with frequent features of mononeuritis multiplex) occurs in about 57 % of patients (Guivellin et al. 1999; Mahr 2009; Chung and Seo 2010). Frequently affected nerves include the peroneal, ulnar, and median nerves. Cranial nerves are rarely involved (Vrancken and Said 2013). MPA may develop insidiously with nonspecific constitutional signs and symptoms or may be disguised as polymyalgia rheumatica.
13.1.3.5 Henoch–Schönlein Purpura (HSP)
Henoch–Schönlein purpura (HSP) is a systemic vasculitis of unknown etiology that involves the small vessels, most notably those in the skin (palpable purpura), gastrointestinal tract, and glomeruli, accompanied by arthralgia or arthritis. HSP is seen predominantly in children and its main histopathological features are leukocytoclastic vasculitis (LCV) mainly in papillary dermis associated with the deposition of IgA immune complexes in small vessels. Peripheral nerves are rarely affected (Mathew et al. 2007; Linskey et al. 2012). The prognosis of HSP is excellent as progressive renal disease occurs only in a minority of patients.
13.1.3.6 Behçet’s Disease (BD)
Behçet’s disease (BD) is a systemic disorder of unknown etiology. The criteria for diagnosis require the presence of oral ulceration plus any two of the following manifestations: genital ulcerations, papulopustular lesions, erythema nodosum-like nodular lesions, positivity of skin pathergy reaction, and uveitis (Walker et al. 1990, Melikoglu et al. 2008). BD also features a systemic vasculitis with a predilection to affect large veins and arteries including vena cava and aorta and its branches. Pseudoaneurysm formation in affected vessels is common. Involvement of small vessels is rare and manifestation of mononeuritis multiplex is exceptional (Takeuchi et al. 1989; Walker et al. 1990).
13.1.3.7 Connective Tissue Diseases
Vasculitis is suspected when a patient with multisystem disease develops symptoms and signs of peripheral nerve dysfunction. However, nerve biopsies in many such patients show nonspecific changes and normal vessels (Olney 1992). Clinicopathological review of our material revealed that non-vasculitic neuropathy-associated with systemic inflammatory disease (NASID) was the fourth most common diagnosis (Table 1.2), and the histological correlates of this included any combination of axonal, demyelinating, and inflammatory changes (Table 1.3). It is impossible to determine whether this represents a sampling error or whether a non-vasculitic pathologic process is at work.
Vasculitis and vasculitic neuropathy are typically seen in rheumatoid arthritis (RA) only after many years of disease activity, in the presence of erosive joint disease, cutaneous nodules, and high titers of rheumatoid factor (Scott et al. 1981; Hawke et al. 1991). Vasculitic neuropathy can rarely precede the diagnosis (Peyronnard et al. 1992; Chang et al. 1984). The development of vasculitis in a patient with RA indicates a poor prognosis (Vollertsen et al. 1986; Hawke et al. 1991). In rheumatoid arthritis a mild predominantly sensory neuropathy is more frequent than the more severe sensorimotor neuropathy associated with vasculitis (Olney 1992). In rare biopsied cases the underlying pathology has been mild axonal loss with segmental demyelination in the absence or paucity of vascular changes, but whether this is primary or secondary demyelination has not been determined (Weller et al. 1970; Beckett and Dinn 1972). Patients suffering from rheumatoid arthritis are also prone to developing other types of neuropathy including entrapments (e.g., median mononeuropathy at the wrist and digital nerve compression secondary to tenosynovitis and arthritis of the carpal bones and digits (Pallis and Scott 1965; Gwathmey et al. 2014). The most devastating type of neuropathy is a progressive multifocal neuropathy, with features similar to those of PAN.
Sjögren syndrome is an autoimmune disease caused by inflammation of the salivary and lacrimal glands. Patients develop the sicca complex (dry eyes and dry mouth), and peripheral neuropathy may be the presenting manifestation (Mellgren et al. 1989; Peyronnard et al. 1992). The clinical picture is usually not a mononeuritis multiplex, but rather a distal sensorimotor neuropathy. Pure sensory neuropathy is also seen and has features suggestive of dorsal root ganglionitis (Pavlakis et al. 2012). Vasculitis (with no other systemic manifestations) has been reported to account for about 15 % of Sjögren-related neuropathies. Important to the diagnosis of Sjögren syndrome is the presence of SS-A (Ro)/SS-B (La) antibodies (Theander and Jacobsson 2008). In Sjögren syndrome a pure sensory neuropathy is occasionally seen and is likely due to spinal ganglionitis (Chap. 21)
Peripheral neuropathy develops in about 10–20 % of patients with systemic lupus erythematosus (SLE) (Chalk et al. 1993; Richardson 1982; Wallace and Metzger 1993; Collins and Periquet 2008; Florica et al. 2011). A distal sensorimotor polyneuropathy, a mononeuritis multiplex, and rarely a CIDP-like picture (Richardson 1982; Rechthand et al. 1984) may be seen. The neuropathy is usually seen after the disease is well established, but can be the presenting manifestation (McCombe et al. 1987; Hughes et al. 1982). The vasculitis affects small vessels and has leukocytoclastic characteristics. PAN-like necrotizing vasculitis may be observed in medium-sized blood vessels. In SLE, CIDP has been infrequently reported (Rechthand et al. 1984; Richardson 1982). Some of the vasculitides encountered in mixed connective tissue disease are similar to the morphologic vascular changes in SLE-associated vasculitis.
Peripheral neuropathy (excluding trigeminal sensory neuropathy and carpal tunnel syndrome) is present in 1–10 % of patients with Scleroderma (Olney 1992; Lee et al. 1984; Averbuch-Heller et al. 1992; Dierckx et al. 1987; Hietaharju et al. 1993). Histological data is scanty, but there are several reported cases of biopsy-proven vasculitic neuropathy in progressive systemic sclerosis (PSS), most often in the presence of Sjögren syndrome (Oddis et al. 1987; Dyck et al. 1987; Vincent et al. 1985). A neuropathy in which vasculitis cannot be implicated, and which can precede generalized clinical manifestations, may be seen in PSS (Di Trapani et al. 1986). Several histological reports have documented prominent epineurial and perineurial collagenization and microangiopathic but non-vasculitic changes similar to those seen systemically with this disease (Richter 1954; Di Trapani et al. 1986; Corbo et al. 1993).
13.1.4 Other Vasculitides
A peripheral neuropathy is said to occur in as many as 14 % of patients with giant cell (temporal) arteritis and frequently precedes the diagnosis by several months (Caselli et al. 1988). Isolated reports exist documenting necrotizing vasculitis, with or without giant cells, involving neural vessels of all sizes in cases of clear-cut temporal cell arteritis (Bridges et al. 1989; Torvik and Berntzen 1968; Merianos et al. 1983; Pons et al. 1987; Nesher et al. 1987). Sites involved include brachial plexus, peroneal nerve, mononeuropathies, and polyneuropathies. Histopathologic features consist of transmural lymphohistiocytic inflammation in the absence of fibrinoid necrosis. Multinucleated giant cell formation occurs associated to degeneration of internal elastica.
A group of nonsystemic, localized vasculopathies termed diabetic lumbosacral radiculoplexus neuropathy and nondiabetic lumbosacral radiculoplexus neuropathy (LRPM) are characterized by intense pain and weakness in the thighs and progressing to affect lower extremities including feet and toes. The clinical course is monophasic but results in protracted morbidity (Gwathmey et al. 2014). Histopathological studies revealed reduction in the number of nerve fibers, perineurial thickening, neuroma formation, neovascularization, and half of the cases featured changes suggestive of microvasculitis.
A heterogeneous group of diseases termed hypersensitivity vasculitis or cutaneous small-vessel vasculitis may present as an idiopathic condition, or secondary to infections, adverse reaction to pharmaceuticals, in the setting of malignancy or autoimmune diseases. Peripheral neuropathy occurs only rarely, except in patients suffering from autoimmune disorders. The hypersensitivity vasculitis that results from the administration of heterologous sera has received the most interest in the past (Iqbal and Arnason 1984).
Paraneoplastic Vasculitic Neuropathy. The most common paraneoplastic vasculitis is leukocytoclastic vasculitis, 75 % of which are caused by hematological malignancies. Second in frequency is small-cell carcinoma of the lung followed by malignancies of the colon, breast, and kidney (Solans-Laqué et al. 2008). In a review of 14 cases, nine cases involved microvasculitis and five cases involved necrotizing vasculitis in medium-sized vessels (Oh 1997; Naka et al. 1991; Choi et al. 2013; Paul 1996).
The most frequently described neuropathy in chronic hepatitis C is a vasculitic neuropathy in the setting of essential cryoglobulinemia (Ferri 2008; Gwathmey et al. 2014; Ramos-Casals et al. 2006). Peripheral neuropathy is more common in essential mixed cryoglobulinemia, but reports of pathological studies are rare (Vrancken and Said 2013).
Vasculitic neuropathy is rare in sarcoidosis, but it is well documented (Said et al. 2002; Vital et al. 2008), including epineurial necrotizing vasculitis in the vicinity of granulomata. Our experience with this disease indicates the presence of naked granuloma in perivascular spaces of epineurial medium-sized vessels, with intrusion into the vessel wall. Some patients suffering from sarcoidosis exhibit the clinical syndrome of mononeuritis multiplex.
13.2 Pathology
13.2.1 General Considerations: Sensitivity of Biopsy
Nerve biopsy is critical for the diagnosis of vasculitis in two clinical situations: “atypical” presentation of systemic vasculitis masquerading as a cryptogenic polyneuropathy and isolated vasculitis of the peripheral nervous system.
The sensitivity of nerve biopsy in the detection of vasculitis is not known because of the variability of selection criteria in published series and the absence of a “gold standard” against which to measure the biopsy. In a study which identified 35 consecutive patients with mononeuritis multiplex from a hospital EMG laboratory, 11 had a previously known rheumatic disease, 9 had the simultaneous onset of systemic disease, and 15 showed only peripheral nerve manifestations (Hellmann et al. 1988). Biopsies showed definite vasculitis in 3 of 5 patients strongly suspected of having polyarteritis nodosa clinically, but in none of 7 biopsies in patients with isolated PNS disease. In the latter group, however, nerve “infarction” was seen in 5 of 7. Dyck et al. (1987) noted that of 45 patients with a biopsy-proven systemic vasculitic illness with neuropathy, nerve biopsy was positive in 58 % in patients, suggestive in 29 %, and nondiagnostic in 23 %. It is possible to find necrotizing vasculitis in an electrically normal nerve (Kissel and Mendel 1992), as has been our experience with 3 patients who underwent nerve and muscle biopsy for suspected systemic vasculitis despite normal nerve conductions studies and EMG. However, the yield is lower than in clinically or electrically involved nerves (Wees et al. 1981). Such data do not make possible estimation of the sensitivity of sural nerve biopsy for the diagnosis of vasculitic neuropathy.
Epineurial vascular involvement usually predominates over endoneurial involvement and is often exclusive; thus, fascicular biopsy is not appropriate when vasculitis is suspected (Dyck et al. 1972; Oh 1990); this is the strongest argument against the utility of fascicular nerve biopsy for diagnostic purposes.
If muscle biopsy can be added to the procedure, the yield may increase an additional 15–45 % (Hawke et al. 1991; Vincent et al. 1985; Dyck et al. 1987). In one series of 83 patients who underwent nerve and muscle biopsy, necrotizing arteritis in the muscle alone was found in 45 %, as compared with 20 % in the nerve alone, and 30 % in both, including patients with seemingly isolated PNS vasculitis (Said et al. 1988). We thus advocate combined nerve and muscle biopsy whenever vasculitis is being considered.
13.2.2 Some Pathological Considerations
Perivascular inflammation is a common and nonspecific finding in peripheral nerve pathology and should be differentiated from “vasculitis.” Throughout this book, the term “vasculitis” indicates inflammation of the vessel and evidence of destruction such as fibrinoid necrosis, thrombosis, hemorrhage, or disruption of the endothelium (Fig. 13.1a, b). Transmural inflammation accompanied by karyorrhexis has substantially the same value as fibrinoid necrosis (Fig. 11.2), whereas the presence of leukocytes within the vessel wall is suggestive but not diagnostic of vasculitis. At times, what seems to be prominent perivascular cuffing around small vessels (Fig. 13.2) has been called “microvasculitis” by some (Oh et al. 1991; Vincent et al. 1985), but in our experience this lesion does not have the same diagnostic specificity as necrotizing vasculitis. For example, some of the patients reported by Leger et al. (1988) as showing neural “vasculitis” likely suffered from CIDP. Vasculitis can involve small-sized endoneurial and epineurial vessels for which the designation of microvasculitis is preferred. The anatomy of peripheral nerve vasculature is reviewed in Chap. 6.
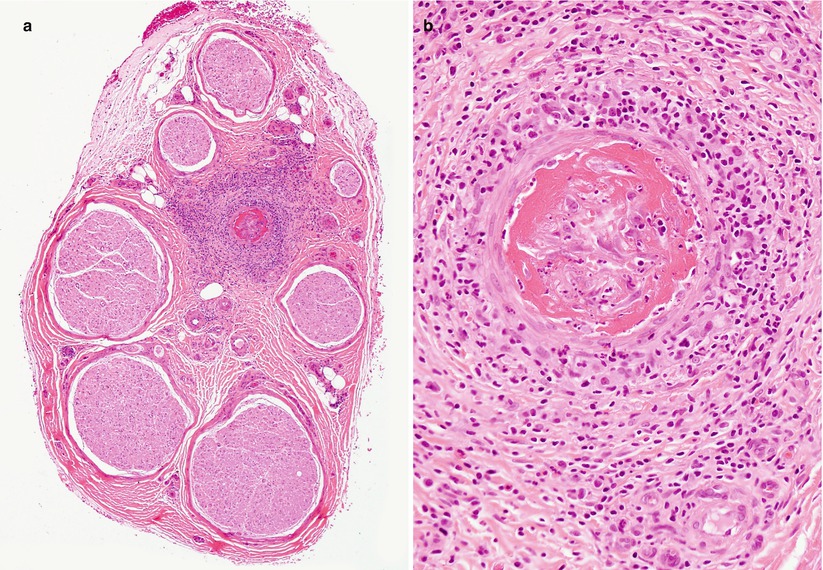
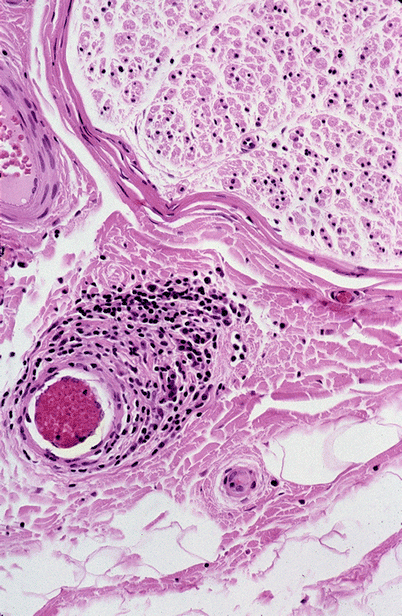
Fig. 13.1
Vasculitic neuropathy: (a) recent fibrinoid necrosis and thrombosis of an epineurial vessel, with perivascular hemorrhage. (b) Transmural inflammation and karyorrhexis are associated with perivascular accumulation of polymorphonuclear leukocytes and mononuclear cells (b) (paraffin, H&E stain; magnification: a, 100×; b, 400×)
Fig. 13.2
Epineurial perivascular inflammation in the absence of necrosis does not meet the gold standard of angionecrosis, although additional adjacent sections may meet that criterion (paraffin, H&E, 400×)
13.2.2.1 Light Microscopy
The hallmark of acute vasculitis is destruction and disorganization of muscularis and endothelial layers of the vessel, with deposition of fibrinoid material in the presence of transmural mononuclear or polymorphonuclear inflammatory cells and thrombosis. The damage is often focal, involving only a segment of the vessel wall (Fig. 13.3a, b). Hemorrhage into the surrounding tissue may be seen (Fig. 13.4a), sometimes in a perineurial or subperineurial crescentic pattern. Perl’s ferrocyanide stain (Fig. 13.4b) will highlight old hemorrhages (Adams et al. 1989), but the specificity of this finding is uncertain (Winer et al. 1992). The use of MSB or PTAH staining (Fig. 13.5a, b) may bring out inconspicuous fibrinoid change. The pattern of involvement in vasculitis is often patchy, with unscathed vessels and nerve fascicles adjacent to severely damaged ones (Fig. 13.6a, b). Epineurial vessels, predominantly arterioles, are much more frequently damaged than endoneurial vessels (Fujimura et al. 1991). The size of vessels affected has diagnostic implication (Table 13.2). Step sections encompassing the entire thickness of the tissue block may be necessary to arrive at the correct diagnosis if initial examination shows nonspecific features such as lymphocytes in the form of large aggregates or perivascularly (Fig. 13.2). The internal elastic lamina is fragmented, and the use of elastic stains alternating with H&E is helpful in the search for vascular damage.
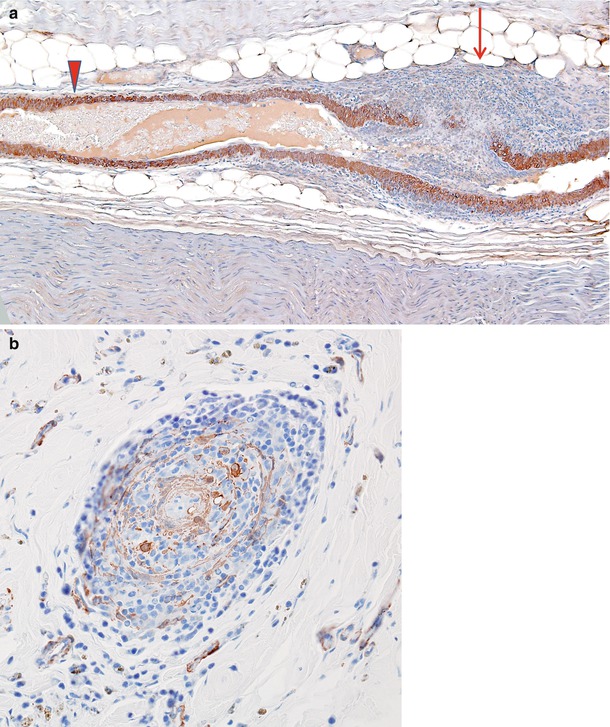
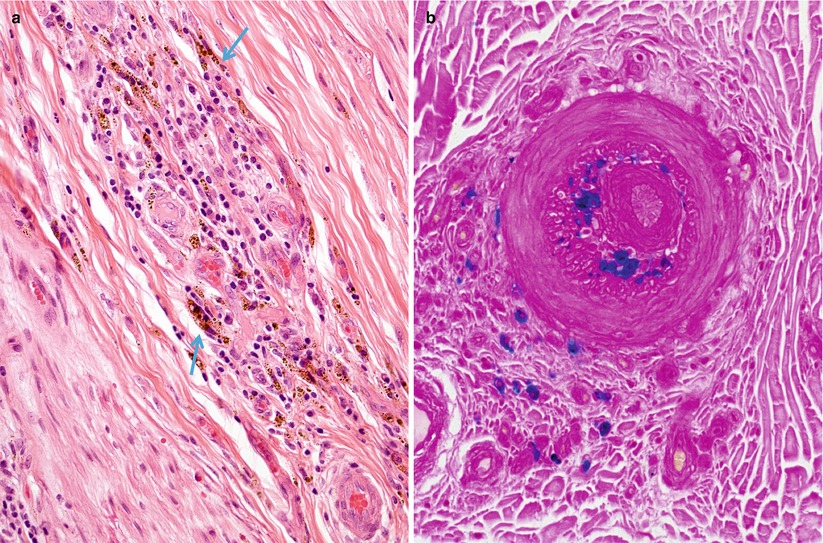
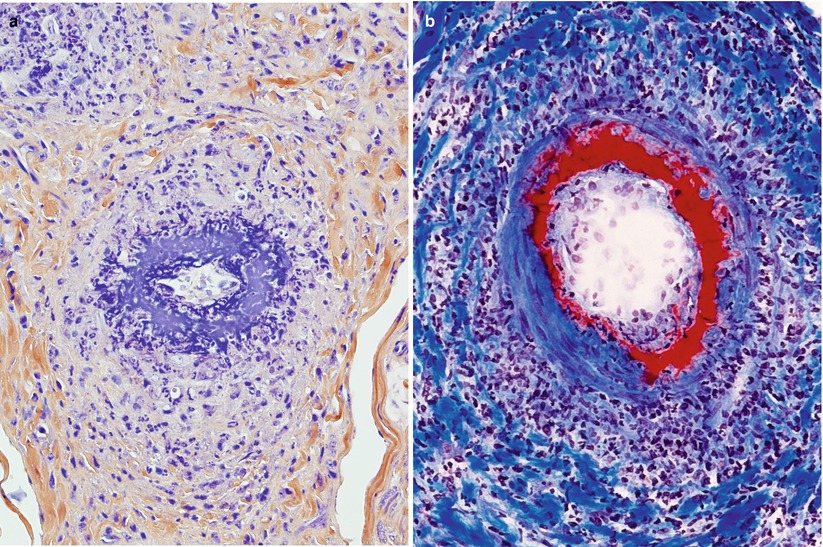
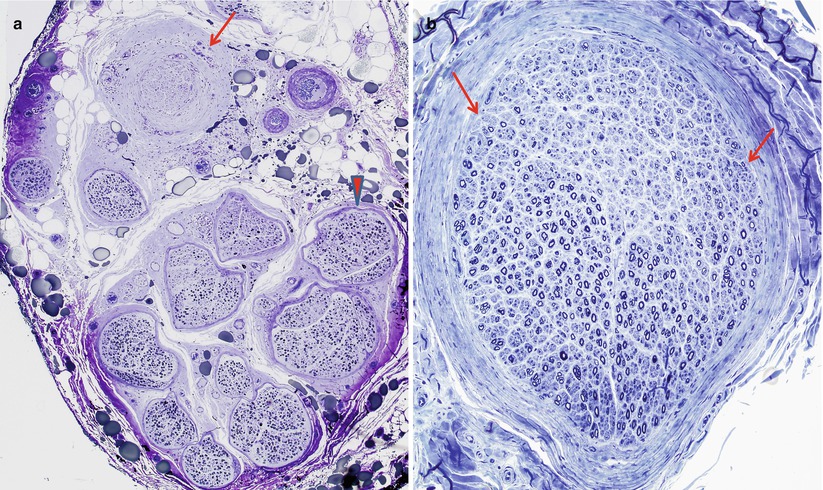
Fig. 13.3
(a) Vasculitis: longitudinal section of epineurial artery shows the focal nature of vasculitis. At the distalmost aspect shown (arrow) marked inflammation has resulted in complete thrombosis and mural destruction only a few hundred microns from a portion of the vessel exhibiting only a perivascular lymphocytic infiltrate (arrowhead). (b) Vascular cross section shows destruction of the entire smooth muscle component of the vessel wall (paraffin, anti-smooth muscle actin (anti-SMA) immunohistochemistry; magnification: a, 100×; b, 200×)
Fig. 13.4
Vasculitis, hemorrhagic residua: perivascular inflammation and hemosiderin deposition (arrows, a) are highlighted by Prussian blue stain (b) for iron (paraffin, a, H&E; b, Perls’ iron stain; magnification: a, b, 200×)
Fig. 13.5
(a, b) Fibrinoid necrosis/fibrin deposition in vasculitis (a) phosphotungstic acid (PTAH) stain; B, Landrum stain for fibrin (magnification: a, b ~400×)
Fig. 13.6
Ischemic pattern of axon loss: (a) typical pattern of fascicle-to-fascicle variability of axon loss is characterized by marked axon loss (arrow) compared to more modest loss (arrowhead). Note that none of the vessels in the cross section demonstrate vasculitis. (b) Individual fascicles show intrafascicular variability with patches of axon loss (delimited by arrows) (1 μ thick plastic sections; magnification: a, 100×; b, 200×)
Table 13.2
Clues to the presence of remote vasculitis
Luminal narrowing or thrombosis |
Disorganization of vessel: intimal hyperplasia, thinning or proliferation of the media, breakup of circumferential ring of internal elastic lamina |
Vessel sclerosis, recanalization |
Proliferation of epineurial capillaries |
Old hemorrhage in nerve (hemosiderin, positive Perl’s Prussian blue) |
Nonuniform fascicular or multifocal axonal degeneration |
Vascular immunoglobulin and complement deposition |
Focal calcification of or near vessel walls |
Focal perineurial damage with aberrant regenerating nerve bundles |
In the acute stage of vasculitis, polymorphonuclear leukocytes can be prominent (Fig. 13.1b and 13.7a–e), but usually T lymphocytes predominate in the vessel wall and perivascular area, with variable numbers of macrophages (Kissel et al. 1989). Any nerve biopsy showing extravasation of neutrophils should be regarded as suspicious for vasculitis, as polymorphonuclear infiltrates are almost never seen in any other cause of neuropathy. However, neutrophils may also be seen in any biopsied tissue if an inordinate amount of time is taken to perform the procedure (30 min or more) (Fig. 7.13). The presence of inflammatory cells within the vessel wall is also suggestive but not diagnostic of vasculitis (vide supra). The inflammatory cells may include epithelioid elements, which can be loosely clustered, palisading, or tightly packed in association with multinucleated giant cells. Eosinophils commonly and plasma cells less frequently contribute to the cellularity. Two of four patients with Churg–Strauss angiitis whose nerve biopsies we have studied had large numbers of eosinophils within the vessel wall in perivascular spaces of epineurium (Fig. 13.8a–e). Conspicuous in one case were Charcot–Leyden crystals within the inflammatory infiltrate (Fig. 13.8d, e). The inflammatory damage to the vessel usually results in narrowing and thrombotic occlusion (Figs. 13.5b and 13.8c). Accumulation of mucoid “edema” in endoneurium, subperineurium, and around vessels is occasionally seen.
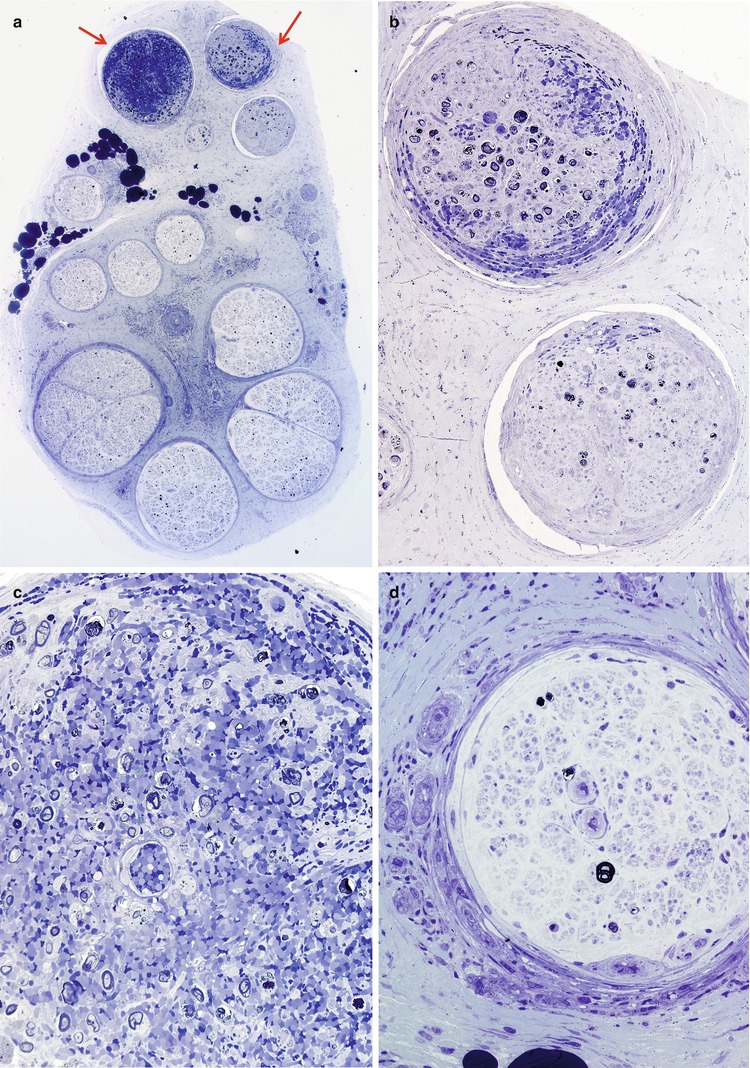
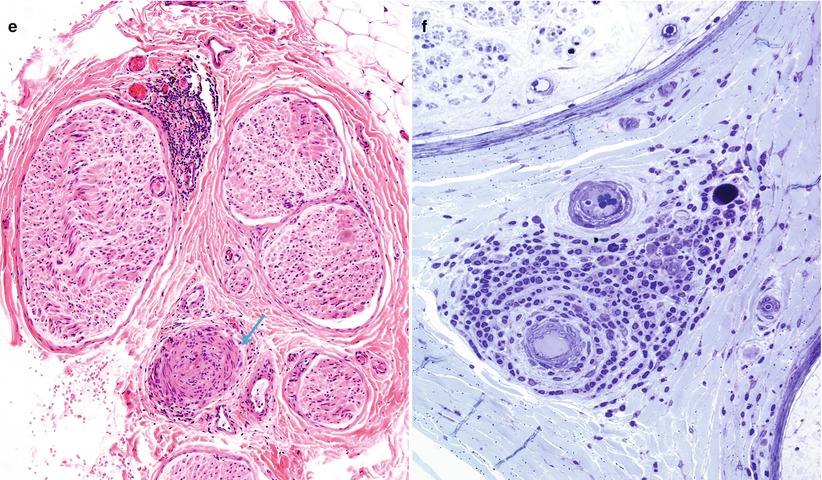
Fig. 13.7




Polyarteritis nodosum (PAN): (a) two fascicles show hemorrhagic necrosis (arrows). (b, c) Higher magnification of involved fascicles show hemorrhage and axonal degeneration. (d) Axonal depletion is the cardinal feature in all other fascicles. (e) Epineurial perivascular chronic inflammation is accompanied by a vessel showing resolving vasculitis (arrow). (f) Perivascular inflammation in the absence of angionecrosis is also common. (a–f 1 μ thick plastic sections, f paraffin, H&E; magnification: a, 40×; b, 200×; c, d 600×; e, 100×; f, 400×)
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
