Fig. 13.1
ACTH-producing PanNET. The tumor cells grow in solid sheets (H&E, ×200)
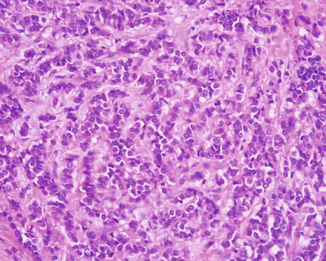
Fig. 13.2
ACTH-producing PanNET with a trabecular pattern of growth and moderate stromal fibrosis (H&E, ×200)
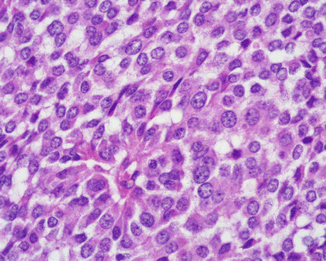
Fig. 13.3
ACTH-producing PanNET. Tumor cells show moderately abundant, eosinophilic, granular cytoplasms and round to oval nuclei with regular contours, evident nuclear membrane, “salt and pepper” chromatin, and small nucleoli (H&E, ×400)
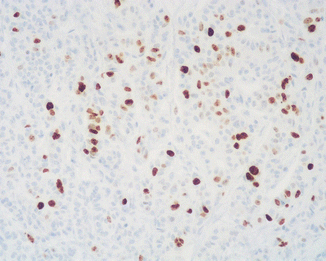
Fig. 13.4
Ki67 immunoreactivity in an ACTH-producing PanNET G2 reveals a proliferative index higher than 3 % (immunoperoxidase and hematoxylin, ×200)
Immunohistochemically, all ACTH-producing pancreatic neuroendocrine tumors react with general neuroendocrine markers (synaptophysin, neuron-specific enolase, chromogranin A, CD56). These tumors are also immunoreactive for ACTH and/or other ACTH-related peptide hormones, such as pro-opiomelanocortin (POMC), melanotropin (MSH), β-endorphin, and met-enkephalin, all identifiable using specific antibodies in at least a fraction of tumor cells [93] (Figs. 13.5 and 13.6). Intriguingly, several neuroendocrine tumors of the pancreas have been reported to show ACTH expression and clinical features of CS only in recurrences or in metastatic localization, while the primary neoplasm was ACTH negative [63, 74]. As it is also clinically evident, plurihormonality is not an infrequent feature of CS-associated PanNETs. Interestingly, a few of these neoplasms have been found to be immunoreactive also for corticotropin-releasing hormone (CRH), including malignant plurihormonal neuroendocrine tumors associated with ZES [74]. In addition, a case of CRH-immunoreactive PanNET associated with ECS, with no expression of ACTH or other hormones, has also been described [69]. All ZES-associated tumors show positive immunostaining for gastrin (Fig. 13.7), whereas insulin was detected in tumors associated with insulinoma syndrome. It has been recently reported that ACTH-producing neuroendocrine tumors of the pancreas can be immunoreactive for CD117 and galectin-3, although the meaning of the expression of these molecules is still unclear [92]. Finally, ACTH-secreting PanNETs show a weak expression of SSRT2A, possibly due to the downregulation of the SSTR by high levels of glucocorticoids, as reported in ACTH-secreting pituitary adenomas [95–97]. However, octreotide therapy has been shown to suppress ACTH and cortisol secretion in several patients with ectopic ACTH production from PanNETs, bronchial carcinoids, and medullary carcinoma of the thyroid [1].
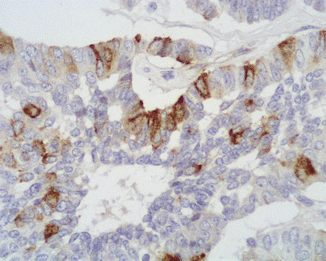
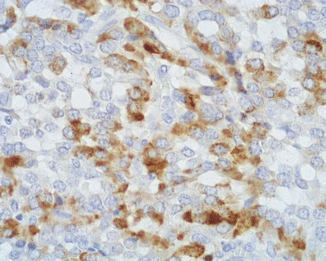
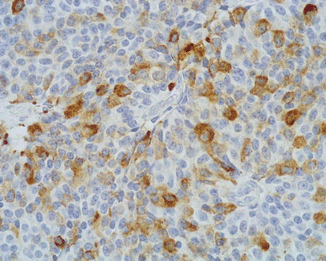
Fig. 13.5
ACTH immunoreactivity is observed in a fraction of neoplastic cells in a Cushing’s syndrome-associated PanNET (immunoperoxidase and hematoxylin, ×400)
Fig. 13.6
This ACTH-producing PanNET is also immunoreactive for β-endorphin (immunoperoxidase and hematoxylin, ×400)
Fig. 13.7
Gastrin immunoreactivity in an ACTH-producing PanNET associated with Cushing’s syndrome and Zollinger-Ellison syndrome (immunoperoxidase and hematoxylin, ×400)
ACTH-producing PanNETs have a high metastatic rate, with 79 % of all cases presenting with systemic disease or developing distant metastases during the history of the disease [91]. As a consequence, the majority of these tumors have a high stage at diagnosis, according to the ENETS system [98]. The liver is the most frequent site of hematogenous localization, and it is almost invariably involved in metastatic disease. Less frequently, distant metastases may occur in the lung, in the bones, and in pelvic organs, i.e., ovaries [91].
13.5 Prognosis
ACTH-producing PanNETs are aggressive tumors, with a median overall survival (OS) of 30 months and a 5-year OS of 25 % [91]. These figures are significantly worse than that of other PanNET subtypes (5-year survival of 97 % for insulinomas, 72 % for gastrinomas, 75 % for somatostatinomas, and 80 % for nonfunctioning PanNETs [see specific chapters in this book]). The association of an ECS and a ZES due to a PanNET in the same patient significantly worsens the patients’ outcome. If this group of patients is excluded from the general ACTH-producing PanNETs population, the median and the 5-year OS rise to 50 months and to 35 %, respectively. By contrast, the association with insulinoma syndrome, as well as the detection of the production of other hormones, does not seem to produce a different prognosis.
References
1.
Beuschlein F, Hammer GD (2002) Ectopic pro-opiomelanocortin syndrome. Endocrinol Metab Clin N Am 31:191–234CrossRef
2.
3.
Del Castillo EB, Trucco E, Manzuoli J (1950) Maladie de Cushing et cancer du pancréas. Presse Med 58:783
4.
5.
6.
7.
8.
9.
Marks V, Samols E, Bolton R (1965) Hyperinsulinism and Cushing’s syndrome. Br Med J 1:1419–1420PubMedCentralPubMedCrossRef
10.
11.
Geokas MC, Chun JY, Dinan JJ et al (1965) Islet-cell carcinoma (Zollinger-Ellison syndrome) with fulminating adrenocortical hyperfunction and hypokalemia. Can Med Assoc J 93:137–1343PubMedCentralPubMed
12.
13.
Burkinshaw JH, O’Brien D, Pendower JE (1967) Cushing’s syndrome associated with an islet-cell tumour of the pancreas in a boy aged 2 years. Arch Dis Child 42:525–531PubMedCentralPubMedCrossRef
14.
Uei Y, Kim U, Itatsu Y (1968) An autopsy case of islet-cell carcinoma with Cushing’s syndrome. Acta Pathol Jpn 18:333–343PubMed
15.
17.
Frederick WC, Gross L (1972) Surgical correction of functioning islet cell carcinoma of the pancreas associated with hyperadrenocorticism. Am J Gastroenterol 57:146–151PubMed
18.
21.
22.
23.
Craig ID, Nelson PG (1975) Pancreatic islet-cell tumour associated with Cushing’s syndrome. Report of a case with estimation of tumour ACTH content. Ulster Med J 44:68–70PubMedCentralPubMed
24.
Imura H, Matsukura S, Yamamoto H et al (1975) Studies on ectopic ACTH-producing tumors. II. Clinical and biochemical features of 30 cases. Cancer 35:1430–1437PubMedPubMedCrossRef
Related posts:
 Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
 Nonfunctioning Pancreatic Neuroendocrine Neoplasms (Including PP-Producing and Calcitonin-Producing Tumors)
Nonfunctioning Pancreatic Neuroendocrine Neoplasms (Including PP-Producing and Calcitonin-Producing Tumors)
 Insulinoma
Insulinoma
 Hyperplastic and Microadenomatous Pancreatic Neuroendocrine Lesions
Hyperplastic and Microadenomatous Pancreatic Neuroendocrine Lesions
 Classification and Staging of Pancreatic Neuroendocrine Neoplasms
Classification and Staging of Pancreatic Neuroendocrine Neoplasms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


