and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Amyloidosis is among the best known of the gain-of-toxic-function protein misfolding diseases. They are characterized by the accumulation and aggregation of nonfunctional and toxic proteins that damage cells and tissues (Blancas-Mejia and Ramirez-Alvarado 2013). Amyloid is detected as an extracellular proteinaceous substance that appears amorphous by light microscopy and fibrillar under the electron microscope. Amyloid fibrils differ from other protein aggregates because they show apple green-yellow birefringence when stained by Congo red and viewed with polarized light. Amyloid consists of beta-pleated sheet micelles forming matted linear non-branching fibrils, 7–10 nm in width. Each fibril comprises two filamentous subunits that run in parallel and twist about each other. The common constituents of all amyloid types are amyloid P component, an endogenous glycoprotein produced by the liver, and various mucopolysaccharides (Pepys 1986, 1990). Different amyloid types are distinguished by their major protein, which is the largest constituent of the deposit. To date there are 30 known extracellular proteins that fulfill the criteria for the designation of amyloid (Sipe et al. 2012), of which five affect the peripheral nervous system (AmTTR, AApoA, AGel, AL, and APrP). Amyloid is insoluble and highly resistant to proteolysis. Its gradual accumulation in a variety of locations ultimately results in clinical disease by mechanisms that have not been fully elucidated.
Amyloidosis can be divided into primary, secondary, familial, and tissue-specific forms (see Adams 2001; Buxbaum 2004; Plante’-Bordeneuve and Kerschen 2013). Primary (AL) amyloidosis refers to amyloid whose major protein is derived from immunoglobulin light chains, predominantly the V region (Feiner 1988). AL implies the presence of plasma cell dyscrasia, whether obvious or occult. Patients with familial amyloidosis have an inherited tendency for deposition of amyloid in various tissues, very often with prominent involvement of peripheral nerve, as seen in several of familial amyloid polyneuropathies (FAPs). Secondary amyloidosis, with serum protein A as the major protein, may occur as part of a chronic systemic inflammatory disease and does not ordinarily cause a clinical polyneuropathy (vide infra). Although there are other forms of amyloidosis (Kyle and Dyck 1993), including one associated with dialysis where the major protein is beta-2 microglobulin and carpal tunnel syndrome is seen (Gertz and Kyle 1989), these have little relevance to peripheral polyneuropathy and nerve biopsy and are not considered further. In 2013 Mead et al (2013) reported a novel prion disease phenotype that is associated with chronic diarrhea and hereditary sensory and autonomic neuropathy caused by a novel PRNP mutation. Deposition of prion protein amyloid was demonstrable systemically and in peripheral nerves.
15.1 Clinical Manifestations
15.1.1 Primary Amyloidosis
Primary amyloidosis is usually a multisystem disease with neural involvement in 20–34 % of patients, and 7–12 % of all primary amyloidosis patients present with an isolated neuropathy months to years before other manifestations of the systemic disease are evident (Buxbaum 1992; Duston et al. 1989; Gertz and Kyle 1989; Trotter et al. 1977). Age at diagnosis of peripheral nerve involvement and disease duration are 57.9 ± 8.7 years and 14.7 ± 7.2 months, respectively (Matsuda et al. 2011). The polyneuropathy is length dependent with a sensory-dominant impairment, early involvement of lower limbs, loss of all sensations, and late development of motor weakness, painful paresthesia, and frequent autonomic dysfunction. This pattern has many similarities to the polyneuropathy manifested by patients afflicted with mTTR amyloidosis.
Traditionally, once amyloid deposition has been documented, the diagnosis of primary amyloidosis is made when evidence for a plasma cell dyscrasia is found, most commonly in the way of a paraprotein. This approach is prone to error because patients with a familial amyloid neuropathy, where no family history is found in as many as 50 % (Li et al. 1992), are as likely as anyone else to have an incidental paraprotein. Systemic amyloid light chain (AL) amyloidosis, the most common systemic amyloidosis, is characterized by a plasma cell disorder in which deposits of the N-terminal region of light chains cause progressive organ failure. More importantly a serum or urine paraprotein is absent in 13 % or more of patients with primary systemic amyloidosis and in almost half of patients with primary amyloidosis presenting with an isolated neuropathy (Buxbaum et al. 1990; Gertz and Kyle 1989). In such cases diagnosis has to be pursued by bone marrow biopsy, skeletal survey, and repeated search for abnormal proteins with immunofixation of serum and urine, and immunoglobulin-free light chain kappa and lambda testing. Monoclonal immunoglobulin-free light chain (Ig FLC) assay is essential in the diagnosis and monitoring of light chain only secreting multiple myeloma (Bence Jones myeloma), and it has allowed the detection of monoclonal protein in some patients with nonsecretory myeloma that were previously undetectable (Uchida et al. 2012). If immunofixation of serum and urine is negative and the Ig FLC (kappa/lambda) ratio is normal (0.26–1.65), AL amyloidosis is unlikely and further evaluation should not be undertaken (Gertz 2013; Lubimova et al. 2012). In Sweden, the median survival time after diagnosis of AL is 3 years.
15.1.2 Hereditary Amyloid Neuropathies
Several distinct familial amyloid polyneuropathies (FAP) have been described, all autosomal dominant. For practical purposes, the FAPs can be classified according to the precursor protein of amyloid (Blancas-Mejia and Ramirez-Alvarado 2013). Clinically, a family history is initially obtained in as few as 50 % (Li et al. 1992), but with intense scrutiny, this may increase substantially (Gertz et al. 1992; Li et al. 1992). Patients typically present with a peripheral neuropathy, although multisystem disease, especially cardiac, is found in many and, uncommonly, there is no clinical nerve involvement at all (Gertz et al. 1992; Ikeda et al. 1987; Meretoja and Teppo 1971; Reilly and King 1993). Three circulating proteins have been identified to date as being the source of a major protein in FAP:
15.1.3 Transthyretin
Hepatocyte-derived transthyretin (TTR) is a serum transport protein, previously called pre-albumin, which serves as a carrier for several substances, including thyroxin and vitamin A. It is encoded by a single copy gene on chromosome 18. Over 113 different missense point mutations in the TTR gene have been associated with deposition of circulating transthyretin as the amyloid major protein (Reilly and King 1993; Plante’-Bordeneuve and Kerschen 2013) in peripheral nerves, heart, gastrointestinal tract, and kidneys. First discovered in 1952 and still the most common pathogenic substitution is the Val30Met mutation (Andrade/Portuguese variant, FAP type I) seen in Portuguese, Japanese, and Swedish patients. Gene penetrance ranges from 69 % in Sweden to 85 % in Portugal (Plante-Bordeneuve and Said 2011). Of interest is the high prevalence of the Val122Ile mutation (most often associated with cardiac disease) in African Americans. Most patients are heterozygous for the TTR mutations, and the amyloid deposits consist of mutant and nonmutant transthyretin. Transthyretin is also synthesized by the retina (retinal pigment epithelium) and choroid plexus, which can lead to vitreous and leptomeningeal accumulations (Coelho et al. 2013)
Although a family history may be negative, it is usually presumed that this is due to incomplete penetrance or inability to examine the entire family. Patients with amyloid neuropathy in which the major protein is TTR and which are truly sporadic (i.e., new mutations in the TTR gene as verified by molecular biological techniques) do seem to exist (Murakami et al. 1992), but their frequency is as yet unknown. In addition normal, nonmutant wild-type transthyretin (wtTTR) is amyloidogenic, and transthyretin amyloid deposits are present in many tissues in at least 10 % of persons over the age of 80 years. This syndrome of systemic senile amyloidosis does not feature deposits in endoneurium or evidence of nerve fibre damage.
In a large study of patients with idiopathic carpal tunnel syndrome, 34 % of tenosynovial tissue obtained at surgery showed amyloid deposition characterized as wtTTR by direct DNA sequencing (Sekijima et al. 2011).
15.1.4 Apolipoprotein A1
Familial amyloid neuropathy (FAP) type III (Van Allen, Iowa variant) is due to amyloid deposits derived from a mutant apolipoprotein A1, encoded on chromosome 11 (Reilly and King 1993). Apolipoprotein A1 is a plasma protein with an extensive α-helical structure synthesized by the liver and the small intestine. The neuropathic pattern of symptoms is associated with the Gly26Arg mutation. The clinical features include length-dependent peripheral neuropathy of variable severity but never prominent, peptic ulcer disease, liver disease, nephropathy, and death from renal failure.
15.1.5 Gelsolin
Gelsolin is an actin-modulating protein encoded on the long arm of chromosome 9q32-34. A point mutation in this protein has been associated with FAP IV (Finland, Meretoja variant, Maury 1991; Sunada et al. 1993). The preferred name is gelsolin amyloidosis (Pihlamaa et al. 2012; Kiuru-Enari and Haltia 2013). The same mutation is seen in unrelated Finnish and Japanese families. A point mutation in the gelsolin gene results in the production of an amyloidogenic circulating degradation product of this protein (Maury and Rossi 1993). Clinically, the neuropathy starts in the third or fourth decade of life and is distinctive by virtue of prominent cranial nerve involvement with facial paralysis, although amyloid deposits are seen throughout the PNS (Meretoja and Teppo 1971) and are accompanied by distinctive corneal lattice dystrophy (this is diagnostic for this disease, sometimes noted years before the appearance of clinical symptoms of neuropathy) and skin changes (cutis laxa) (Kiuru-Enari et al 2002).
15.1.6 “Sporadic” Amyloid Neuropathy
Up to 39 % of patients with amyloid neuropathy have no family history and show no evidence for plasma cell dyscrasia (Dalakas and Cunningham 1986). These may be patients with primary amyloidosis but no detectable paraprotein, or “familial” (usually mTTR) amyloidosis with an incomplete family history, incomplete penetrance, or new mutations (Adams et al. 1992). Such patients pose a clinical challenge in terms of investigation and treatment. Recent advances in immunohistochemistry and Ig-free light chain determination (Lubimova et al. 2012) can play a major role in the management of this group (vide infra).
15.1.7 Clinical Features
The first signs and symptoms of MTTR-FAP develop most commonly from the third to the fifth decade of life (Val30Met variant, patients with other mutations experience a later age of onset). The disease affects both men and women equally (Coelho et al. 2013). In its classical form (Val30Met mutation), patients display two patterns of sensorimotor deficit associated with variable autonomic dysfunction (orthostatic hypotension, bladder or sexual dysfunction, and constipation) and extra-neurological manifestations (nephrotic syndrome or renal failure, hepatomegaly, peripheral neuropathy, orthostatic hypotension, and macroglossia Kyle and Gertz 1995; Plante-Bordeneuve and Said 2011). One pattern consists of a length-dependent sensorimotor polyneuropathy. Symptoms start with discomfort in the feet with impaired thermal sensitivity and decreased pinprick sensation with preservation of light touch, proprioception, and tendon reflexes; the other type begins with focal deficits resulting from intraneural amyloid aggregates followed by a bilateral sensory motor polyneuropathy. An ataxic phenotype was recently reported in France in up to 26 % of TTR-FAP patients (Adams et al 2014). At the outset the patients display foot numbness and imbalance. On examination there is vibration and position sense loss and diffuse areflexia. The course is rapid with progressive ataxia. Patients are often misdiagnosed as having CIDP. Different TTR mutations (Val93Met) may present with an ALS phenotype (Adams et al 2014).
Once the length-dependent neuropathy is established, it progresses relentlessly proximally to thighs. Motor deficits occur in the same fashion from distal to proximal. Upper limbs are then affected, there is loss of pain sensation distally, and the development of ulcers in feet and foot arthropathy occurs in the course of few years. By electrophysiological tests, in the early stages quantitative sensory testing and sympathetic skin tests confirm small fiber involvement (Heldestad and Nordh 2007). Later, EMG shows active and chronic neurogenic changes. In the natural course, the disease is progressive and invariably fatal after an average duration of 10–13 years.
15.1.8 Treatment
Correct diagnosis of familial amyloidosis is important for both genetic counseling and the avoidance of unnecessary invasive testing. Liver transplantation has been shown to stabilize disease progression because it eliminates the production of mutant protein, particularly if performed early in the disease (Holmgren et al. 1993; Adams 2001; Yamamoto et al. 2007). However, additional deposition of wtTTR on the template of preexisting amyloid occurs after transplantation, leading to cardiomyopathy and worsening of polyneuropathy. Liver transplantation has no effect on ocular complications or eventual CNS symptoms of amyloidosis caused by the persistent synthesis of mTTR by retinal epithelial cells and the choroid plexus (Hara et al. 2010). Tafamidis, a small chaperone molecule that binds to transthyretin and prevents tetramer dissociations into monomers (the rate-limiting step in amyloidogenesis), slows deterioration of peripheral nerve function and improves quality of life (Coelho et al. 2012). New techniques of lowering mTTR gene expression with TTR-specific lipid-based siRNAs are in development (Love et al. 2010). Coelho et al. (2013) reported the use of a lipid nanoparticle envelope to facilitate the safe delivery of the siRNA to hepatocytes, which resulted in dramatic and sustained specific knockdown of transthyretin production. There is no specific treatment for either gelsolin amyloidosis or for apolipoprotein A-1 FAP.
Once the amyloidosis is confirmed to be of light chain origin, treatment options include stem cell transplant or trials of chemotherapy, which include corticosteroids, alkylating agents (melphalan, cyclophosphamide), immunomodulatory drugs (thalidomide, lenalidomide), and proteasome inhibitors (bortezomib) (Gertz 2013). Alternative therapeutic strategies to liver transplant include tafamidis, diflunisal, antisense oligonucleotides, and small interfering RNA (Adams et al 2014).
15.1.9 Secondary Amyloidosis
In general, polyneuropathy is not considered to be a feature of secondary (AA) amyloidosis (Benson et al. 1975). However, there are reported cases of secondary amyloidosis associated with autonomic, cranial, and perhaps sensorimotor neuropathy (Horn et al. 1991; McGill et al. 1986; Nordborg et al. 1973; Tsunoda et al. 1994). The autonomic features can be explained by AA amyloid deposits in sympathetic ganglia (McGill et al. 1986). However, secondary amyloid can also be deposited in the endoneurium (McGill et al. 1986; Tsunoda et al. 1994). Of two such reported instances, no polyneuropathy was seen in the first case (McGill et al. 1986), and the presence of a non-amyloidogenic circulating paraprotein might have explained the neuropathy in the second case (Tsunoda et al. 1994).
15.2 Pathology
15.2.1 Utility of Nerve Biopsy
It is important to emphasize that there are alternatives to nerve biopsy. Biopsy of abdominal fat pad or rectal mucosa is a high yield, low morbidity option if primary or familial amyloidosis is suspected (Gertz et al. 1988, 1992). In one large study skin biopsy was as sensitive as nerve biopsy for detection of amyloid in Portuguese FAP (Guimaraes et al. 1987). Muscle biopsy probably has a higher yield than nerve, at least in primary amyloidosis (Dalakas and Engel 1979); we favor a combined biopsy of muscle and nerve.
In familial amyloid neuropathy nerves from asymptomatic/presymptomatic known carriers may appear normal histologically or show abnormalities similar to those of patients with a neuropathy (Guimaraes et al. 1987). Skin and nerve biopsies detect amyloid in about half of asymptomatic carriers, with skin biopsy being somewhat more sensitive (Leite et al. 1987). This contradicts the results of previous work where no amyloid was found in asymptomatic or presymptomatic carriers (Carvalho et al. 1976; Harats et al. 1989). In symptomatic patients, data regarding sensitivity of the sural nerve biopsy is hard to extract from the literature, because the diagnosis of amyloid neuropathy has historically been based on histological findings in nerve alone. One large study of familial amyloid neuropathy (TTR 30) demonstrated neural amyloid deposits in 42 of 44 clinically affected patients (Guimaraes et al. 1987). The two whose nerve biopsies did not contain amyloid did show moderate–severe fiber loss in a typical pattern (unmyelinated more than myelinated fibers), and their skin biopsies were positive for amyloid. In another study of a variety of familial amyloid neuropathies, 3 out of 23 patients had negative nerve biopsies, but the presence or absence of clinical peripheral nerve involvement was not stated (Gertz et al. 1992). These data suggest a high but not 100 % sensitivity for sural nerve biopsy in clinically affected patients with FAP, and a role for skin or other tissue biopsy if the clinical picture and pattern of fiber loss suggest amyloid neuropathy but no amyloid is noted. Data on asymptomatic/presymptomatic FAP patients suggest that nerve biopsy is not very sensitive in revealing amyloid, as only 23 % of asymptomatic patients with histological neuropathy (i.e., loss of fibers) actually showed amyloid deposits (Leite et al. 1987). Whether this relates to sampling error or the fact that amyloid deposition does not directly cause the neuropathy remains unknown.
The sensitivity of nerve biopsy in primary amyloidosis with peripheral neuropathy has been considered to be high. Amyloid was detected in 86–100 % of sural nerve biopsies (Kelly et al. 1979; Kyle and Dyck 1993), but a selection bias obviously confounds these numbers. In another report, only 2 of 8 nerve biopsies were amyloid positive in patients with a neuropathy consistent with that of amyloidosis and who were ultimately diagnosed as having primary amyloidosis (Simmons et al. 1993). We have seen three patients with systemic amyloidosis (two primary, one indeterminate) and neuropathy in whom sural nerve biopsy did not reveal amyloid, although in one the neuropathy might well have been due to the circulating IgM paraprotein, not amyloid deposition. In another, the diagnosis was first suspected after biopsy showed a selective loss of small myelinated fibers, but abdominal fat pad biopsy was necessary to arrive at the diagnosis, while in the third the diagnosis was revealed at autopsy. Thus, if clinical suspicion is high and nerve biopsy is unrewarding, the clinician should consider another biopsy site.
15.2.2 General Considerations
Our experience with eight cases (3 familial TTR, 3 light chain, and 2 not characterized) and that of some workers (Li et al. 1992) suggests there is no significant difference between the histopathology seen in familial amyloid neuropathy and in primary amyloidosis and the two are discussed together below.
Physical methods (autoclaving) and histochemical techniques (potassium permanganate, alkaline guanidine) exist which can distinguish between some of the different types of amyloid (Elghetany and Saleem 1988), but immunohistochemistry currently represents the most useful method, as it reliably distinguishes familial from light chain amyloid proteins (Linke et al. 1986). The amyloid of FAP and primary amyloidosis is resistant to potassium permanganate while that of secondary amyloidosis is not (Sommer and Schroder 1989). This is of minimal value because secondary amyloidosis does not cause a peripheral polyneuropathy (vide supra).
Using strain-free lenses, amyloid displays birefringence under polarized light even when unstained, but the classic apple-green-yellow birefringence is demonstrated best with Congo red staining in 5–10 μm thick sections. Fresh-frozen sections exhibit the highest affinity for Congo red. A number of non-amyloid substances stain positively with Congo red but show no birefringence (fibrin, elastic fibers, mast cell granules) while others even show the birefringence (fungi, plant cell walls, cotton fibers, starch). For details and additional technical considerations, see Elghetany and Saleem 1988; Francis 1990. The sensitivity of Congo red staining for amyloid is excellent, but the affinity for this stain is gradually lost with prolonged formalin fixation. We have seen cases where small amyloid deposits were detected only on EM, presumably due to sampling error, and if clinical suspicion is high, one should not abandon the search for amyloid if Congo red staining is negative. The use of fluorochromic dyes (thioflavin T, thioflavin S, and Phorwhite BBU) is also a reliable method for the screening of amyloid in fresh-frozen and in paraffin sections (Francis 1990; Waldrop et al. 1972).
Renaut bodies have been mistaken for amyloid deposits because of their amorphous light microscopic appearance and the presence of fibrillar material intermixed with collagen on EM. The microfibrillar component, typically slightly wider (8–13 nm vs. 7–10 nm) and not as long and straight as amyloid fibrils, is probably oxytalan, a constituent of elastic fibers (Ghadially 1988a; Weis et al. 1993) (Fig. 15.1a, b). The absence of Congo red staining and the characteristic location and shape of Renaut bodies should clarify the situation. Similar microfibrils are often seen in subperineurial, endoneurial interstitial, and a perivascular location and should not be mistaken for amyloid. There should be no difficulty distinguishing endoneurial “edema,” which is Congo red negative and Alcian blue positive, from amyloid.
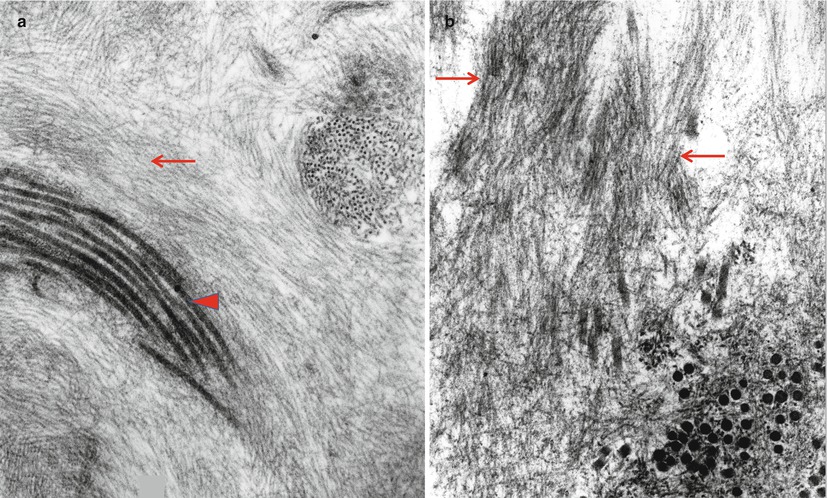
Fig. 15.1
The ultrastructural differences between oxytalan (arrow, a), collagen (arrowhead, a), and amyloid fibrils (arrows, b) are the slightly greater thickness of oxytalan and the apparent rigidity of the amyloid fibril as compared with curved or wavy appearance of oxytalan (a, b: 65,100×)
15.2.3 Light Microscopy
Amyloid accumulates in epineurium, perineurium, and endoneurium. One case we examined showed amyloid deposition mostly in the epineurial fat and could have been missed with a fascicular biopsy. When seen in the endoneurial interstitial compartment, amyloid may be found in large pools or distributed diffusely (Figs. 15.2, 15.3a–c, and 15.4a–d). Amyloid masses can be seen focally in the vessel wall or forming perivascular collars (Figs. 15.4a–d and 15.5). In the latter case the resulting appearance may be dismissed as nonspecific hyaline thickening of the vessel unless Congo red-stained sections are examined routinely under polarized light. It has been suggested that in primary, as contrasted with familial, amyloidosis there is a tendency for the deposits to be more prominent in and around vessels, particularly the vasa nervorum in the epineurium (Asbury and Johnson 1978), but we have not observed such a distinction (Li et al 1992). Perineurial and subperineurial deposition is common and often prominent (Figs. 15.3c and 15.4c, d). In secondary amyloidosis there may be epineurial, but not endoneurial amyloid deposition (Belokrenitzky 1911). Inflammatory cells are not a feature of amyloid neuropathy.
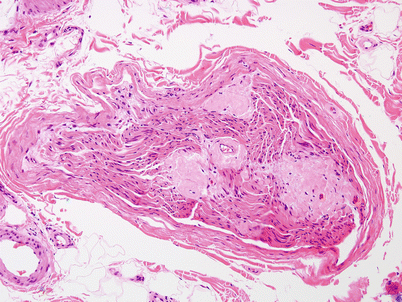
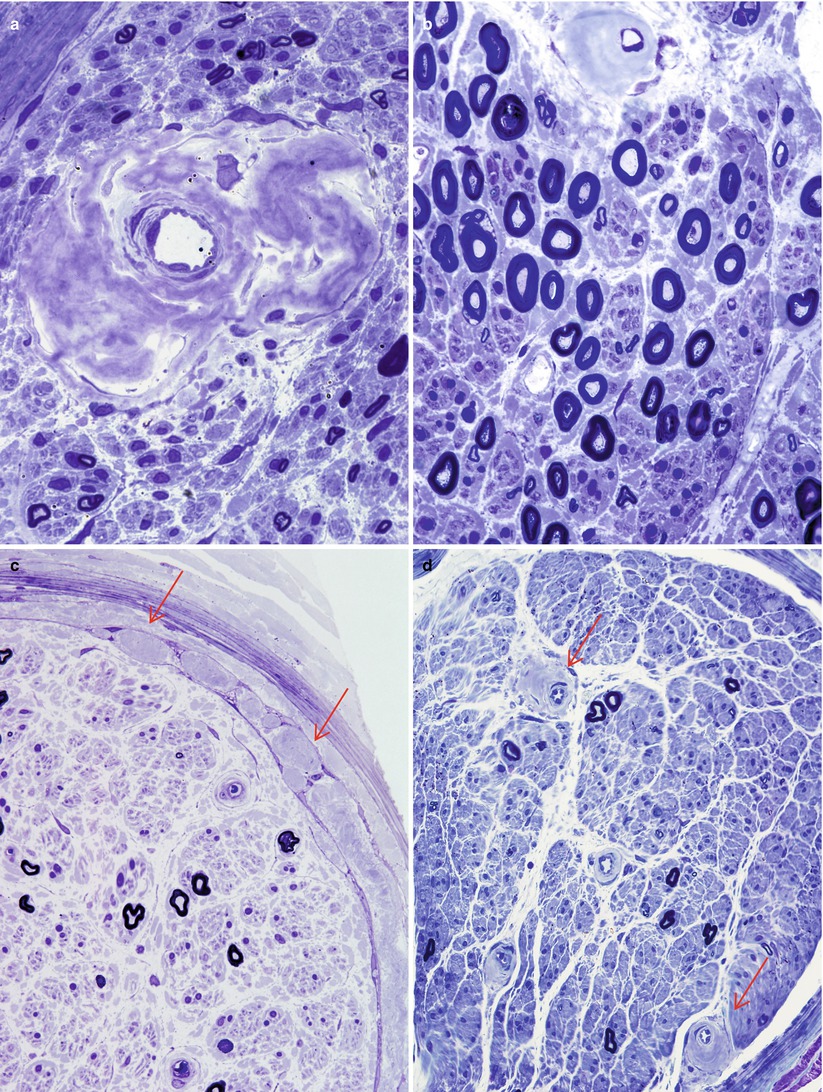
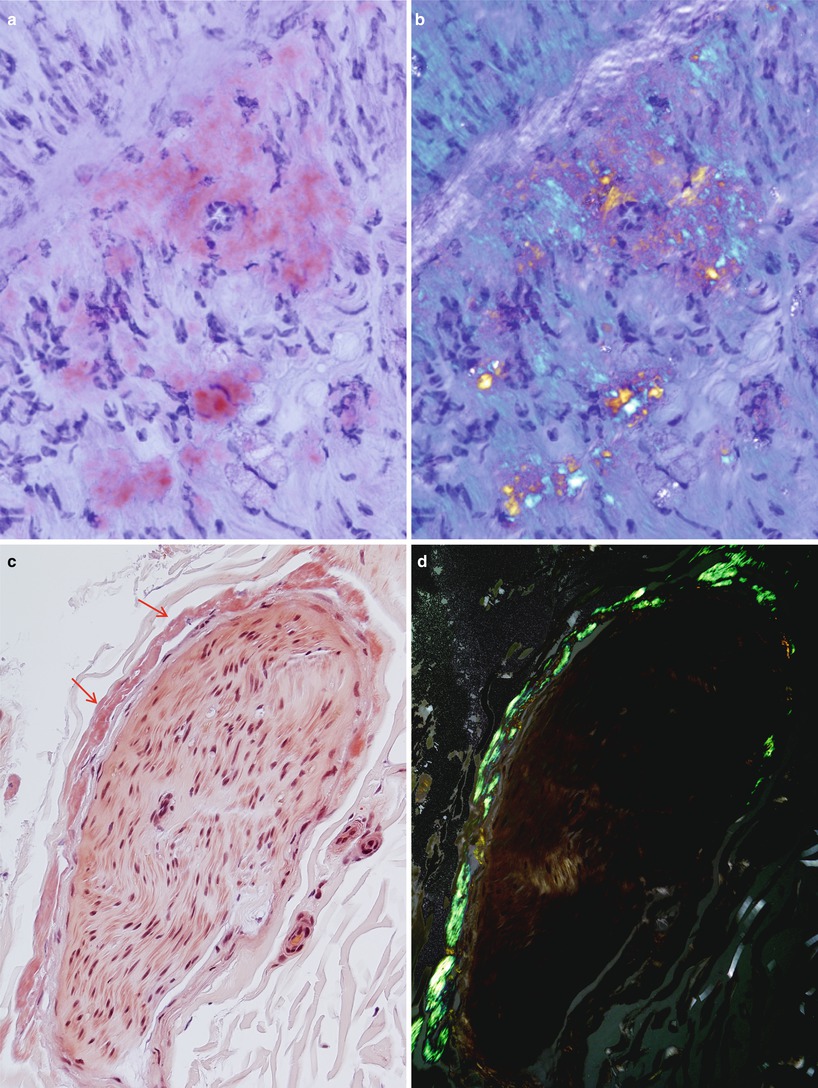
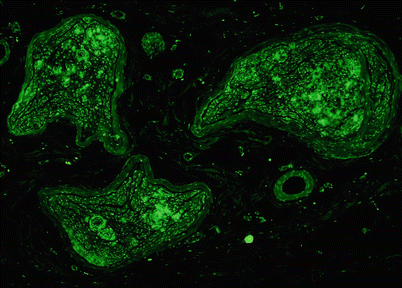
Fig. 15.2
Amyloidosis: clumps of pink amorphous amyloid are found within the endoneurium, adjacent to the perineurium and perivascularly (H&E, paraffin)
Fig. 15.3
Amyloidosis: (a) irregular perivascular clumps of amyloid. (b) Although large myelinated axons appear well maintained in this plastic section, small myelinated axons are greatly diminished in number. Note again the asymmetry of amyloid deposition around the vessel. (c) Expansion of the subperineurial and intraperineurial spaces by clumps of amyloid (arrows, c). (d) More modest irregular perivascular aggregates (arrows) (a–c: 1 μ thick toluidine blue-stained plastic section, 1,000×; D 400×)
Fig. 15.4
Amyloidosis: delicate wispy amyloid expands the endoneurium as shown by Congo red (CR) stain (a) which, upon polarization, results in apple-green fluorescence (b). Perineurial deposition is demonstrated by CR staining (arrows, c) and with polarization (d) (paraffin sections, a–d 400×)
Fig. 15.5
A large endoneurial accumulation of amyloid is demonstrated with thioflavin S fluorescence (paraffin, 200×)
The predominant pathological process is axonal degeneration. The degree of fiber loss depends on the evolution of the disease. In early stages there is usually selective loss of unmyelinated and small myelinated fibers, but as the disease progresses, fibers of all sizes are severely affected (Fig. 15.3a–c). Sometimes the selective loss of unmyelinated fibers is very striking (Fig. 15.3b) (Dyck and Lambert 1969). A spectrum ranging from moderately active Wallerian to chronic axonal degeneration with regenerating clusters may be seen, although we have not found the latter to be prominent in our material. Although amyloid neuropathy is typically axonal, several authors have reported the presence of segmental myelin changes using teased fibers (Dyck and Lambert 1969; Hanyu et al. 1989; Jedrzejowska 1977; Said et al. 1984; Thomas and King 1974). We have never seen prominent segmental demyelination in amyloid neuropathy.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


