Amyotrophic Lateral Sclerosis and Motor Neuron Diseases
Amyotrophic Lateral Sclerosis and Motor Neuron Diseases
Rebecca Traub
Hiroshi Mitsumoto
INTRODUCTION
Several different diseases are characterized by progressive degeneration and loss of motor neurons in the spinal cord with or without similar lesions in the motor nuclei of the brain stem, the motor cortex, or both and by replacement of the lost cells by gliosis. All these can be considered motor neuron diseases (plural). The term motor neuron disease (singular), however, is used to describe an adult disease, amyotrophic lateral sclerosis (ALS), in which both upper and lower motor neurons are affected. (The terms motor neuron disease and amyotrophic lateral sclerosis have become equivalent in the United States.)
The term spinal muscular atrophy (SMA) refers to syndromes characterized solely by lower motor neuron signs. By conventional usage, the term spinal muscular atrophy is reserved for the child-hood form, which is heritable, as described in Chapter 141.
Other motor neuron disease variants include progressive muscular atrophy, in which patients show only lower motor neuron signs; primary lateral sclerosis, in which patients show only upper motor neuron signs; progressive bulbar palsy, with weakness limited to bulbar muscles; and monomelic amyotrophy, in which the usual lower motor neuron findings are restricted to a single limb. These motor neuron disease subtypes are described more in detail in the following sections (Table 85.1).
It should be noted that there is some clinical overlap between motor neuron diseases, pure motor neuropathies (particularly those that are hereditary), and the hereditary spastic paraplegias (HSP). Distinguishing and classifying clinical phenotypes that lie in the overlap between these diagnoses often clinically challenging at present because of lack of diagnostic biomarkers.
TABLE 85.1 Motor Neuron Disease Variants
Amyotrophic lateral sclerosis (ALS)
Upper and lower motor neuron degeneration
Progressive muscular atrophy (PMA)
Purely lower motor neuron involvement
Primary lateral sclerosis (PLS)
Purely upper motor neuron involvement
Progressive bulbar palsy
Bulbar symptoms only or bulbar-onset ALS
Monomelic muscular atrophy
Lower motor neuron predominant, one arm
Bibrachial amyotrophy
Lower motor neuron predominant, both arms
AMYOTROPHIC LATERAL SCLEROSIS
DEFINITION
ALS is a disease of unknown cause and pathogenesis. It is defined pathologically as one in which there is degeneration of both upper and lower motor neurons. Charcot made the key clinical and pathologic descriptions, and the disease is named for him in Europe. In the United States, the disease is colloquially called Lou Gehrig disease after the famous baseball player who had the disease. Clinically, ALS is defined by evidence of both lower motor neuron disease (weakness, wasting, and fasciculation) and upper motor neuron disease (spasticity, hyperactive tendon reflexes, Hoffmann signs, Babinski signs, or clonus) in the same limbs. The accuracy of clinical diagnosis is assumed to be more than 95%, but that figure has not been formally tested. Nevertheless, the reliability of clinical diagnosis suffices to make the findings in the history and examination part of the definition.
EPIDEMIOLOGY
ALS is found worldwide in roughly the same prevalence (about 50 × 10-6), with the exception of a few geographic areas with high prevalence of ALS, parkinsonism, and dementia complex, most notably on the island of Guam in the past. Case-control studies have not identified consistent risk factors related to occupation, diet, or socioeconomic status. There appears to be an increased risk of ALS among certain professional athletes and some suggestion that repeated head trauma may increase the risk for the disease.
The disease is generally of middle and late life. Only 10% of cases begin before age 40 years; 5% begin before age 30 years, with a greater proportion of those younger cases attributed to hereditary motor neuron diseases. An increase in age-adjusted incidence is seen in succeeding decades, except for a decrease after age 80 years. In most series, men are affected one to two times more often than women. There is no known ethnic predilection except slightly higher incidence in Norway because of some concentration of genetic mutations.
GENETICS
Approximately 5% to 10% of ALS cases are familial. Most are autosomal dominant in inheritance, but autosomal recessive and X-linked mutations have also been described. Increasingly recognized are “sporadic” ALS cases, which can be linked to specific genetic mutations, sometimes as a result of de novo mutations or incomplete penetrance. New mutations accounting for familial ALS continue to be described, and at the time of publication of this book, new mutations are likely to have been found not included in this chapter.
The two most common forms of autosomal dominant familial ALS are related to mutations in the C9ORF72 gene and the SOD1 gene. C9ORF72 repeat expansions are linked to both ALS, frontotemporal dementia (FTD), and often a combination of both clinical phenotypes. C9ORF72 mutations are currently the most common cause of familial ALS. Mutations in the SOD1 gene (ALS1) are the second most common cause of familial ALS. Many mutations in this gene have been described, and clinical phenotype is variable. Evidence suggests that mutant protein exerts its effects through accumulation of toxic protein. The SOD1 mutation has been used extensively in research as a model for studying pathology in ALS and identifying compounds for treatment trials.
TABLE 85.2 Familial Amyotrophic Lateral Sclerosis
Name
Site
Gene
Inheritance
Comment
FTD-ALS
9p21
C9ORF72
AD
ALS and FTD
Hexanucleotide expansion
Most common cause of familial ALS
ALS1
21q22.1
SOD1
AD
Adult
15%-20% of familial ALS
ALS2
2q33
Alsin
AR
Juvenile; may resemble PLS
ALS3
18q21
—
—
—
ALS4
9q34
Senataxin (SETX)
AD
Juvenile; slow progression; allelic to CMT2
ALS5
15q15.1-q21.1
SPG11
AR
Juvenile
Most common AR ALS
Also seen in HSP
ALS6
16p11.2
Fused in sarcoma (FUS)
AD
Adult
ALS7
20p13
—
AD
Adult
ALS8
20q13.33
VAPB
AD
Adult
ALS9
14q11.2
ANG
AD
—
ALS10
1p36.22
TARDBP
AD
Some with FTD
ALS11
6q21
FIG4
AD
Allelic to CMT 4J (AR)
ALS12
10p13
OPTN
AD/AR
—
ALS13
12q24.12
ATXN2
AD
CAG expansions and increased ALS risk
Expansions also seen with SCA2
ALS14
9p13.3
VCP
AD
Mutations also seen with IBMPFD
ALS15
Xp11.21
UBQLN2
X-linked
Reduced penetrance in women
ALS16
9p13.3
SIGMAR1
AR
Juvenile
ALS17
3p11.2
CHMP2B
AD
May cause FTD
ALS18
17p13.2
PFN1
AD
—
ALS19
2q34
ERBB4
AD
—
ALS20
12q13.13
HNRNPA1
AD
May cause multisystem proteinopathy
ALS21
5q31.2
MATR3
AD
Formerly distal myopathy 2, VCPDM
Other genes described and implicated in familial ALS-DAO, NEFH, HNRNPA2B1, SQSTM1.
FTD, frontotemporal dementia; ALS, amyotrophic lateral sclerosis; AD, autosomal dominant; AR, autosomal recessive; PLS, primary lateral sclerosis; HSP, hereditary spastic paraplegia; CMT 2, Charcot-Marie-Tooth hereditary neuropathy type 2; CMT 4J, Charcot-Marie-Tooth neuropathy type 4J; CAG, cytosine-adenine-guanine; SCA 2, spinocerebellar ataxia type 2; IBMPFD, inclusion body myopathy with Paget disease and frontotemporal dementia; VCPDM, vocal cord and pharyngeal dysfunction with distal myopathy.
Other familial ALS gene mutations have provided insight into potential pathogenic mechanisms in sporadic disease. Mutations in the fused in sarcoma (FUS) gene (ALS6) and TAR DNA-binding protein 43 (TDP-43) gene (ALS10) suggest a possible mechanism of RNA regulation and metabolism underlying sporadic disease. The pathogenic expansion in C9ORF72 familial ALS may also exert effects through RNA toxicity.
A list of the currently identified mutations in familial ALS at the time of publication of this text are listed in Table 85.2.
Although not typically included among the list of familial ALS, X-linked recessive spinobulbar muscular atrophy (Kennedy disease) should be included as a hereditary motor neuron disease. The gene maps to Xq11-q12, the site of the androgen receptor. The mutation is an expansion of a cytosine-adenine-guanine (CAG) repeat. Symptoms usually begin after age 40 years, with dysarthria and dysphagia with prominent tongue and mentalis fasciculations and with a slow course and limb weakness delayed for years. Upper motor neuron signs are often absent. In contrast to ALS, there is often an associated large-fiber sensory peripheral neuropathy. Gynecomastia is present in most but not all patients. Diagnostic clues include the characteristic distribution of signs, lack of upper motor neuron signs, slow progression, and a family history suggesting X-linked inheritance.
PATHOBIOLOGY
The cause of sporadic ALS is not known. The only established risk factors are age and family history. Environmental factors have been suspected, likely in combination with genetic susceptibility, but no clear risk exposures have been identified. There is increasing interest in repetitive head trauma and a link to chronic traumatic encephalopathy, although this link has yet to be well established. Theories regarding the cause of ALS have included infectious causes, including retroviruses, effects of excitotoxic amino acids, mitochondrial dysregulation, oxidative stress, and autoimmune causes, but little evidence conclusively supports one of these causes. Increasing evidence points to abnormal RNA processing and metabolism underlying ALS, although the exact mechanisms and triggers have yet to be elucidated.
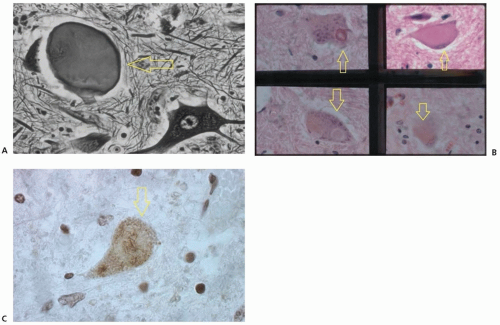
FIGURE 85.1 Pathologic findings in ALS. A: Axonal swelling and axonal spheroids (arrow) (Bodian silver stain, ×400 magnification). B: Motor neuron degeneration (arrows) (hematoxylin and eosin stain, ×400 magnification). C: Ubiquitinated skein-like inclusion (arrow) (ubiquitin immunoperoxidase stain, ×400 magnification).
PATHOLOGY
The pathology of ALS implies selective vulnerability of motor neurons, which show several neuronal inclusions that include ubiquitinated skeins or Lewy-like formations and Bunina bodies (Fig. 85.1). These structures are found in most patients with sporadic ALS. In some familial forms, a different form is the “hyaline conglomerate,” which includes neurofilaments and does not contain ubiquitin. Authorities believe the cellular abnormalities identify a common basic mechanism for the syndromes of ALS, progressive muscular atrophy, primary lateral sclerosis, and amyotrophic lateral sclerosis-frontotemporal dementia (ALS-FTD).
The neuronal antigen in the inclusions recognized by anti-bodies to ubiquitin has been identified as TDP-43, and mutations in the TDP-43 gene are responsible for approximately 5% of familial ALS.
CLINICAL MANIFESTATIONS
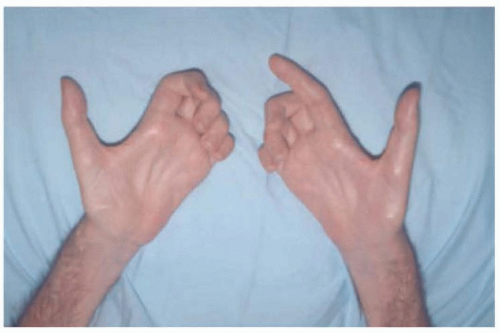
Weakness may commence in the legs, hands, proximal arms, or bulbar muscles (with dysarthria and dysphagia). Often, the hands are affected first, usually asymmetrically, and with atrophy (Fig. 85.2). Progressive painless weakness is the typical primarily clinical feature of ALS. Gait is impaired because leg muscles are weak, and footdrop is characteristic, although proximal limb muscles are sometimes affected first. Alternatively, a spastic gait disorder may ensue. Slowly, the weakness becomes more severe, and more areas of the body are affected, leading to an increasing state of dependency. Muscle cramps (attributed to the hypersensitivity of denervated muscle) and weight loss (resulting from the combination of muscle wasting and dysphagia) are characteristic symptoms. Respiratory impairment is usually a late symptom but may, rarely, be an early or even the first manifestation; breathing is compromised by diaphragm and paresis of intercostal muscles, and dysphagia may lead to aspiration and pneumonitis, both of which can be the terminal event. Sensation is not clinically affected unless a preexisting neuropathy is present; pain and persistent paresthesias are atypical for this diagnosis, unless there is a complicating disease (e.g., diabetic neuropathy). Typically, bladder function is spared. The eye muscles are affected only exceptionally. Pain is not an early symptom but may occur later when limbs are immobile due to severe spasticity and joint contracture.
FIGURE 85.2 Hand atrophy in ALS.
Lower motor neuron signs must be evident if the diagnosis is to be considered valid. Fasciculations may be seen in the tongue, even without dysarthria. If there is weakness and wasting of limb muscles, fasciculations are almost always seen. Tendon reflexes may be increased or decreased; the combination of overactive reflexes with Hoffmann signs in arms with weak, wasted, and fasciculating muscles is virtually pathognomonic of ALS. Unequivocal signs of upper motor neuron disorder are spasticity, Hoffmann or Babinski signs, and clonus. If a spastic gait disorder is seen without lower motor neuron signs in the legs, weakness in the legs may not be found, but incoordination is evident by clumsiness and slowness in the performance of alternating movements (Table 85.3).
Only gold members can continue reading. Log In or Register to continue