Vasculitis affecting the central or peripheral nervous system often presents major diagnostic and therapeutic challenges for physicians because of its variable clinical manifestations and the lack of specific diagnostic tests other than biopsy. Central nervous system (CNS) vasculitis comprises a heterogeneous group of inflammatory diseases that affect leptomeningeal and parenchymal blood vessels of the brain with different but frequently overlapping clinical and pathologic manifestations.
The histopathologic changes of CNS vasculitis essentially consist of an inflammatory infiltrate within the vessel wall associated with necrosis, vessel occlusion and infarction, and in some cases, hemorrhage. The cases of CNS vasculitis are protean: It may occur as an idiopathic primary autoimmune process or within the context of systemic to autoimmune disease, as a reaction to an infection such as varicella-zoster or a neurodegenerative process such as amyloid angiopathy, after drug exposure (e.g., cocaine), as a manifestation of radiation exposure, and in the setting of malignancy. Although the precise pathogenesis often remains obscure, in all cases, the immune system plays a central role, and immunosuppressive agents are the cornerstones of treatment.
There are several different classifications for CNS vasculitis. Some classifications are based on the size of the affected vessels (small, medium, large), whereas other classifications are based on histologic features (e.g., granulomatous, lymphomatous, or leukocytoclastic inflammation) or immunologic markers (e.g., the association with antineutrophil cytoplasmic antibodies, or ANCAs, with some forms of vasculitis). CNS vasculitis can also be further broadly classified into primary and secondary vasculitis depending on whether the process is confined to the CNS or is part of a systemic illness.
In 2012, the International Chapel Hill Consensus Conference proposed a revised nomenclature system that groups different vasculitides by taking in consideration multiple factors including size of vessels and whether the vasculitis involves a single organ or is part of a more systemic condition (Table 42.1).
PRIMARY ANGIITIS OF THE CENTRAL NERVOUS SYSTEM
Primary angiitis of the central nervous system (PACNS) is associated with select inflammation and destruction of small- and medium-sized arteries of the brain parenchyma, spinal cord, and leptomeninges, resulting in symptoms and signs of CNS dysfunction. The term angiitis is synonymous with vasculitis and refers to involvement of blood vessels on both the arterial and venous sides of the circulation. PACNS is also frequently referred to as primary central nervous system vasculitis.
EPIDEMIOLOGY
The annual incidence rate of PACNS is 2.4 cases per 1 million person-years. The disease affects patients of all ages, peaking at 50 years of age, with a 2:1 male predominance.
PATHOBIOLOGY
The etiology and pathogenesis of PACNS is unknown. Infectious agents such as herpes zoster virus, West Nile virus, and varicellazoster virus have been proposed as possible causes or triggers.
Three main histopathologic patterns are generally seen: granulomatous, lymphocytic, and necrotizing vasculitis. In lymphocytic PACNS, immunohistochemical staining reveals predominant infiltration by memory T cells in and around small cerebral vessels, signifying an antigen-specific immune response occurring in the wall of cerebral arteries.
CLINICAL FEATURES
Due to diffuse involvement of the CNS, the clinical manifestations are variable and nonspecific, with a clinical course ranging from hyperacute to stepwise to chronic and insidious. Mental obtundation and decreased cognition are the most frequent clinical presentations, usually evolving subacutely as a progressive encephalopathy. Other symptoms may include headaches, seizures, and focal neurologic deficits due to ischemic stroke or cerebral hemorrhage. Signs and symptoms of systemic vasculitis, such as peripheral neuropathy, fever, weight loss, or rash, are by definition lacking in PACNS.
DIAGNOSIS
On the basis of clinical experience and evidence from published work, Calabrese and Mallek proposed a set of criteria for the diagnosis of PACNS; the diagnosis is established when all three of the following criteria are met (Table 42.2).
There is no specific laboratory diagnostic test for PACNS, although routine laboratory testing, serologic evaluations, lumbar puncture results, neuroimaging studies, cerebral angiography, and brain biopsy may all have roles in the evaluation (Table 42.3). Acute phase reactants such as the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are usually normal in PACNS; elevated levels should raise suspicion of systemic involvement by either an infectious, malignant, or inflammatory process. Both serologic testing and analyses of the cerebrospinal fluid (CSF) are important for excluding secondary causes of CNS dysfunction that may mimic PACNS. Nonspecific abnormalities, primarily leukocyte and protein elevations, are seen on CSF analysis in 80% to 90% of patients with pathologically documented disease.
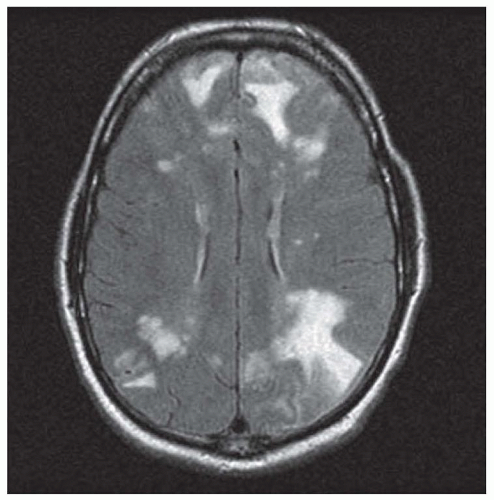
Magnetic resonance imaging (MRI) typically shows multifocal small-vessel infarcts and hemorrhages of varying age (Fig. 42.1). These vascular lesions often involve structures that are not typically involved in conventional cerebrovascular disease, such as the corpus callosum. In some cases, large-vessel infarcts, convexity subarachnoid hemorrhage, or small foci of gadolinium enhancement can occur. It is extremely rare for PACNS to cause advanced disease with severe symptoms with a normal MRI.
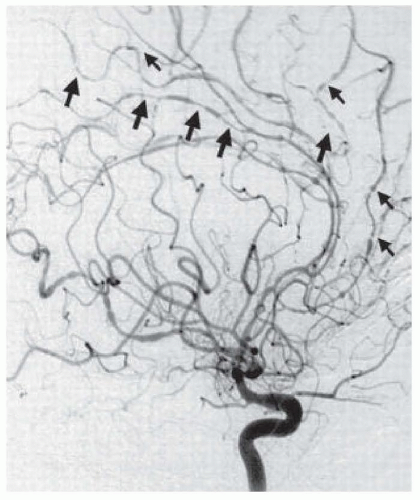
The gold standard for the detection of vascular abnormalities is digital subtraction angiography. In PACNS, cerebral angiography may show areas of ectasia and stenosis referred to as beading circumferential or eccentric vessel irregularities with sharp cutoffs, delayed arterial emptying, small arteriovenous anastomotic channels, and rarely, microaneurysms (Fig. 42.2). The main condition that PACNS must be differentiated from a patient with beading on diagnostic angiography is reversible cerebral vasoconstriction syndrome (RCVS), a much more benign condition discussed later in this chapter (see also Chapter 43). About 4% of patients with primary CNS vasculitis present with a solitary tumorlike mass lesion due to brain tissue inflammation. Given the poor resolution of computed tomography (CT) and magnetic resonance (MR) angiography for distal medium and small blood vessels, these tests have limited sensitivity for diagnosing PACNS.
TABLE 42.1 Vasculitis Syndromes: 2012 International Chapel Hill Consensus Conference
Syndromes
Pathologies
Large-Vessel Vasculitis
Giant cell (temporal) arteritis
Granulomatous arteritis of aorta and its major branches; predilection for extracranial branches of carotid artery; often involves temporal artery; patients older than 50 years with polymyalgia rheumatica
Takayasu arteritis
Granulomatous arteritis of aorta and major branches; patients younger than age 50 years
Medium-Sized Vessel Vasculitis
Polyarteritis nodosa
Necrotizing inflammation of medium- or small-sized arteries; no glomerulonephritis or vasculitis of arterioles, capillaries, venules
Kawasaki disease
Arteritis of large, medium-sized, small arteries plus mucocutaneous-lymph node syndrome; coronary arteries often involved; aorta and veins may be involved; usually in children
Small-Vessel Vasculitis
Granulomatosis with polyangiitis (GPA) (Wegener granulomatosis)
Granulomatous inflammation including respiratory tract and necrotizing vasculitis of small to medium vessels (capillaries, venules, arterioles, arteries); necrotizing glomerulonephritis common
Eosinophilic granulomatosis with polyangiitis (EGPA) (Churg-Strauss syndrome)
Eosinophil-rich granulomatous inflammation of respiratory tract and necrotizing vasculitis of small- to medium-sized vessels; with asthma and eosinophilia
Microscopic polyangiitis (MPA)
Necrotizing vasculitis with few or no immune deposits; affects small vessels (capillaries, venules, arterioles); may involve small- and medium-sized arteries; common features: necrotizing glomerulonephritis and involvement of pulmonary capillaries
IgA vasculitis (IgAV) (Henoch-Schönlein purpura)
Vasculitis with IgA-dominant immune deposits on small vessels (capillaries, venules, arterioles); affects skin, gut, and glomeruli plus arthritis or arthralgia
Cryoglobulinemic vasculitis
Vasculitis with immune deposits on small vessels (capillaries, venules, arterioles); cryoglobulins in serum; skin and glomeruli often involved
Variable-Vessel Vasculitis (VVV)
Behçet disease (BD)
Vasculitis that can affect arteries or veins accompanied by cutaneous, ocular, articular, gastrointestinal, and/or central nervous system inflammatory lesions
Cogan syndrome (CS)
Arteritis (affecting small, medium, or large arteries), aortitis, aortic aneurysms, and aortic and mitral valvulitis accompanied by ocular inflammatory lesions, including interstitial keratitis, uveitis, and episcleritis, and inner ear disease, including sensorineural hearing loss and vestibular dysfunction
Vasculitis associated with systemic disease
Vasculitis that is associated with and may be secondary to (caused by) a systemic disease (e.g., rheumatoid vasculitis, lupus vasculitis)
Vasculitis associated with probable etiology
Vasculitis that is associated with a probable specific etiology (e.g., hydralazine-associated microscopic polyangiitis, hepatitis B virus-associated vasculitis, hepatitis C virus-associated cryoglobulinemic vasculitis, etc.)
IgA, immunoglobulin A.
Modified from Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1-11
TABLE 42.2 Diagnostic Criteria for Primary Angiitis of the Central Nervous System
• History or clinical findings of an acquired neurologic deficit of unknown origin after a thorough initial basic assessment
• Cerebral angiography reveals classic features of vasculitis, or a CNS biopsy sample shows vasculitis.
• There is no evidence of systemic vasculitis or any other disorder to which the angiographic or pathologic features can be attributed to.
CNS, central nervous system.
TABLE 42.3 Suggested Laboratory Testing for Central Nervous System Vasculitis
Basic Laboratory Testing
Complete blood count with differential
Serum urea nitrogen and creatinine
Serum aspartate and alanine aminotransferases
ESR and C-reactive protein
Urinalysis
CSF Studies
Cell count (mild to moderate CSF pleocytosis)
CSF protein (range 44-1,034 mg/dL) CSF glucose (usually normal)
Oligoclonal bands
Gram stain and cultures
Cytology and flow cytometry when malignancy is suspected
PCR and IgG, IgM directed at infectious sources (below)
Specialized Laboratory Testing
Antinuclear antibodies
Rheumatoid factor
Antibodies to the Ro/SSA, La/SSB, Sm, and RNP antigens
Antibodies to double-stranded DNA
Antineutrophil cytoplasmic antibodies (ANCA)
Serum C3 and C4
Serum cryoglobulins
Serum and urine protein electrophoresis with immune electrophoresis
Infectious Etiologies (in appropriate circumstances as indicated)
Bacterial—Mycoplasma PCR and serology, antistreptolysin O test (ASOT), syphilis serology, Mantoux skin test
Viral—serology for hepatitis B and C, parvovirus B19, HIV, herpes simplex virus, Epstein-Barr virus (EBV), cytomegalovirus (CMV), varicella
Fungal culture (Aspergillus, Coccidioides, and Histoplasma species)
Parasite: serum anticysticercal antibodies demonstrated by immunoblot assay and CSF ELISA for detection of anticysticercal antibodies or cysticercal antigens
Thrombophilia Investigations
Prolonged activated partial thromboplastin time (aPTT), which does not correct with mixing
FIGURE 42.1 Fluid-attenuated inversion recovery (FLAIR) MRI showing multifocal infarcts in a patient with biopsyproven PACNS.
FIGURE 42.2 Typical angiographic findings in a patient with central nervous system vasculitis. Arrows point to areas of alternating stenosis and ectasia. (From Hajj-Ali RA, Ghamande S, Calabrese LH, et al. Central nervous system vasculitis in the intensive care unit. Crit Care Clin. 2002;18:897-914).
Arteries with diameter below 0.4 mm, arterioles, and capillaries are beyond the resolution of conventional angiography, and because PACNS often involves small blood vessels, the sensitivity of angiography in biopsy-proven PACNS cases is only 60%. Thus, a negative angiogram cannot exclude the diagnosis of PACNS. Furthermore, cerebral angiography has limited specificity since not infrequently, patients with angiographic findings interpreted as being positive for vasculitis are not found to have vasculitic changes at brain biopsy. It should be noted that normal biopsy specimens may reflect sample error due to the patchy distribution of inflammation. The false-negative rate of an initial biopsy is 25%; in some cases, two or more biopsies are necessary to establish the diagnosis. The yield of biopsy is improved with larger tissue blocks (i.e., 1 cm3) with inclusion of the overlying meninges and when areas affected with enhancement on MRI are targeted.
TREATMENT
No randomized clinical trials of medical management for PACNS exist. First-line therapy usually starts with glucocorticoids. Fulminant PACNS in hospitalized patients usually starts with empiric intravenous methylprednisolone, 15 mg/kg/day for 3 to 5 days, followed by prednisone, 1 mg/kg/day to a maximum of 100 mg/day. Less severe disease in outpatients can be started on prednisone alone. Glucocorticoids should be continued at the initial dose for 4 to 6 weeks after which a slow taper should be initiated.
Patients with biopsy-confirmed granulomatous variant PACNS should be started on a combination of glucocorticoids and cyclophosphamide. Cyclophosphamide is given in order to induce remission in either a daily oral regimen (1.5 to 2 mg/kg/day) or a monthly intravenous regimen (600 to 750 mg/m2, infused once a month). After induction of remission, typically in 3 to 6 months, cyclophosphamide therapy is switched to maintenance therapy with other agents such as mycophenolate mofetil or azathioprine.
In patients with lymphocytic variant PACNS or those in whom the diagnosis is performed by cerebral angiogram and not confirmed by biopsy, an initial glucocorticoid treatment is only followed by cyclophosphamide if the neurologic decline is progressive. As a general rule, chemotherapeutic immunosuppressant therapy should only be given to patients with biopsy-proven PACNS.
Treatment response is monitored by periodic reassessment of symptoms and neuroimaging abnormalities. A follow-up MRI should be obtained 4 to 6 weeks after beginning treatment, then every 3 to 6 months throughout therapy, and subsequently according to the evolution of the disease to assess for progression of the disease.
OUTCOME
Earlier descriptions characterized PACNS as a fatal disease, and most cases were diagnosed at autopsy. With better understanding of the disease and with aggressive immunosuppression, more favorable outcomes and improving mortality rate have been reported. Recent reports indicate that the short-term mortality rate is 10% and that 20% suffer severe functional impairment. Mild cognitive deficits and reduced energy level remain common among those with a good functional recovery. Approximately 30% of patients experience a relapse of the disease after going into remission, which requires reescalation of immunosuppression.
AMYLOID β-RELATED ANGIITIS
Amyloid β-related angiitis (ABRA) is a form of CNS vasculitis in which perivascular β-amyloid is thought to act as a trigger for inflammation mediated by CD68+ macrophages and CD3+ T lymphocytes. The condition develops in patients with symptomatic or asymptomatic amyloid angiopathy (see also Chapter 38), which presents as dementia, gait disorder, and progressive brain hemorrhages (both microbleeds and parenchymal intracerebral hemorrhage). Patients with ABRA often exhibit altered mental status and respond to immunosuppressive treatment. Common CSF findings include an elevated protein and a lymphocytic pleocytosis. MRI often demonstrates T2 hyperintense lesions extending through the cortical white matter and often gray matter, suggestive of breakdown of the blood-brain barrier and a reversible leukoencephalopathy. Cerebral angiography shows beading in a minority of patients, perhaps due to involvement of exclusively medium- and small-sized vessels. In every case on brain biopsy, microglia, macrophages, and T cells surround amyloid-laden vessels. After initiating antiinflammatory treatment consisting of steroids or cyclophosphamide for a duration ranging from 2 weeks to several months, a majority of patients with ABRA show improvement. However, some patients relapse, and other patients do not improve or progressively decline.
CENTRAL NERVOUS SYSTEM VASCULITIS ASSOCIATED WITH SYSTEMIC VASCULITIS
GIANT CELL (TEMPORAL) ARTERITIS
Giant cell arteritis (GCA), also known as temporal arteritis because of its predilection for superficial temporal artery involvement, is a large-vessel granulomatous vasculitis. The disease usually affects the aorta or its major branches, with a predilection for the branches of the cervical internal and external carotid arteries and the vertebral arteries. The intracranial vessels are generally spared. GCA is often linked with polymyalgia rheumatica in terms of its systemic manifestations, ESR elevation, and response to steroid therapy.
Epidemiology
GCA has an annual incidence that is highest among white individuals, ranging between 10 and 20 cases per 100,000 among persons older than 50 years of age. The incidence is markedly lower in persons of Asian or African descent in the range of 1 case per 100,000. Although GCA almost never occurs before age 50 years, the incidence increases thereafter, reaching its peak incidence between the ages of 70 and 80 years.
Pathobiology
Although the etiology of GCA is unknown, there appears to be an interplay between increasing age, a genetic predisposition, and infectious triggers that involves both the cellular and humoral immune system, resulting in an acute vascular inflammatory state. Current literature regarding the molecular basis of GCA suggests that activated dendritic cells, residing in the vessel wall, play an important role in the pathophysiology by initiating the pathogenic cascade, recruiting T cells and macrophages to form granulomatous infiltrates.
GCA is a panarteritis, including all the arterial layers. Thrombosis may develop at sites of active inflammation. Fibrosis, scarring, and narrowing or occlusion of the arteries develops secondary to the inflammation, tissue injury, and repair process.
Clinical Manifestations
GCA is classically characterized by a combination of five features:
Age older than 50 years,
A localized new-onset headache,
Tenderness over the temporal artery,
An elevated ESR (≥50 mm/h), and
Biopsy revealing a necrotizing arteritis with a predominance of mononuclear cells or a granulomatous process with multinucleated giant cells.
The presence of three of the above five criteria is associated with 94% sensitivity and 91% specificity for the diagnosis of GCA. GCA typically presents with a constellation of constitutional symptoms, headache and tenderness over the temporal artery, jaw claudication, visual symptoms, and symptoms of polymyalgia rheumatica (diffuse myalgias and body aches). Because intracranial vascular involvement is rare in GCA, transient ischemic attacks (TIAs) and stroke are usually secondary to embolism or flow failure related to cervical vertebral or internal carotid lesions.
Neurologic manifestations arise in approximately 30% of patients, with more than half being peripheral neuropathies, followed by one-third with TIA or stroke, one-third with ophthalmologic symptoms, and one-fifth with neuro-otologic syndromes. Ophthalmologic symptoms can include transient monocular blindness (i.e., retinal TIA or amaurosis fugax) due to internal carotid artery stenosis or sustained unilateral visual loss due to anterior ischemic optic neuropathy or occlusion of the central retinal artery (see also Chapter 9).
Only gold members can continue reading. Log In or Register to continue