Parkinson’s disease (PD) is a common adult-onset neurodegenerative disorder whose disabling signs are engendered by the loss of specific subsets of neurons, including the dopaminergic neurons in the substantia nigra. To date, researchers still have a limited understanding of the key molecular events that provoke neurodegeneration in this disease. A prevalent etiologic hypothesis is that PD may result from a complex interaction among environmental toxic factors, genetic susceptibility traits, and aging. As for PD pathogenesis, the current consensus is that the neurodegenerative process results from a combination of both cell-autonomous (i.e., mechanisms that take place within the compromised neuron) and non–cell-autonomous mechanisms (i.e., mechanisms that take place outside the compromised neuron and that contribute to its demise). Putative cell-autonomous mechanisms have been brought to light primarily by the discovery of PD genes, which has led to the hypothesis that processes such as protein misfolding, dysfunction in the ubiquitin–proteasome pathway, alterations in mitochondrial bioenergetics, dysregulation in calcium homeostasis, and impaired turnover of mitochondria may be pivotal in the cascade of deleterious events underlying the demise of dopaminergic neurons. As for the proposed non–cell-autonomous mechanisms, these involve mainly the prion-like behavior of misfolded proteins and neuroinflammation. This chapter discusses the evidence and the ongoing lines of research relevant to the quest of unraveling the cause and mechanism of neuronal death in PD.
DEFINITION AND DIAGNOSTIC CONSIDERATIONS
Over the past decade, PD has garnered widespread interest, in part, because several public figures and celebrities were identified as having this illness. PD is considered the second most common degenerative disorder of the aging brain after the dementia of Alzheimer’s disease. In keeping with this, it is currently estimated that more than a million individuals are affected with PD in the United States alone, and, with the aging character of our society, this figure is forecast to increase dramatically over the coming years. The overall incidence of PD in the United States (which surges upward after the age of 60) is roughly 13.4 per 100,000 individuals/year (1), and is approximately twice as frequent in men as in age-matched women (1); whether PD frequency varies among ethnic groups or geographic location remains an unsettled issue (1).
PD is progressive with a mean age at onset of 55. Although the introduction of effective symptomatic treatments such as levodopa has prolonged survival, PD patients, especially with severe parkinsonism or dementia (2), still exhibit a higher mortality risk compared to healthy controls (3). The mean disease duration of PD, as defined by the period between onset of clinical manifestations and death, is estimated to be 10.1 to 12.8 years (4,5). What remains uncertain is whether the observed shortened life expectancy of PD patients results from the disease per se or from the ensuing motor and cognitive impairments, which increase the odds of accidental fatalities such as falling, aspiration pneumonia, pressure skin ulcers, malnutrition, and dehydration. Relevant to this point is the finding that twice as many PD patients die from pneumonia as in the age-matched control population (4). This is more than a discussion of semantics, as it goes to the heart of how best to care for PD patients.
As illustrated in Fig. 7.1, the main neuropathologic feature of PD is the loss of the neuromelanin-containing dopaminergic neurons of the nigrostriatal pathway (6), which depletes the brain of dopamine (7) and leads to the emergence of motor abnormalities such as tremor at rest, rigidity, slowness of voluntary movement, and postural instability (8). By the time patients become symptomatic, approximately 60% of the nigral dopaminergic neurons have been lost and striatal content in dopamine has been reduced by approximately 80%. It can thus be concluded that disease onset predates the expression of the motor manifestations of PD, which has been estimated to be about 4.5 years by 18F-fluorodeoxyglucose with positron emission tomography (PET) (9). In addition to demonstrating that the presymptomatic period of PD appears to be relatively short, this PET study has also documented that the pattern of glucose metabolic alterations here is not consistent with the idea that the neurodegeneration in this illness is a simple exacerbation of the normal age-related decay of the nigrostriatal dopaminergic system (9). Although the lion’s share of attention is consistently paid to the nigrostriatal pathway, it must be remembered that, in reality, degenerative changes in PD are not restricted to that specific pathway and that as neuropathologic findings are noted in other dopaminergic and nondopaminergic cell groups, including locus coeruleus, raphe nuclei, and nucleus basalis of Meynert (10). This is again important with respect to the proper management of PD, as there are some quite disabling features, such as postural instability and cognitive impairment, depression, or constipation, especially in advanced patients, that may not find their pathophysiology in the damage to the dopaminergic system and usually do not improve with levodopa therapy.
Also of importance is the fact that more than 30 different neurologic syndromes share PD clinical features (Table 7.1). Thus, a definite diagnosis of PD is often achieved only at autopsy and, customarily, relies not only on finding a loss of nigrostriatal dopaminergic neurons but also on the identification of intraneuronal inclusions, or Lewy bodies, which can be seen in many of the surviving cells of all affected brain regions (see Fig. 7.1). Lewy bodies are spherical eosinophilic cytoplasmic aggregates of a fibrillary nature that, as illustrated in Fig. 7.1, are composed of a variety of proteins, including α-synuclein, parkin, ubiquitin, and neurofilaments (11,12). Many authorities, however, question whether identification of Lewy bodies should still be necessary for the diagnosis of PD, in light of the fact that cases of inherited PD linked to parkin mutations typically lack Lewy bodies and are still considered to be cases of PD. These facts raise the question as to whether the current nosology of parkinsonian syndromes needs to be revised to distance itself from a classification typically based on the disease’s clinical and neuropathologic hallmarks to one based on the disease’s molecular characteristics. Conceivably, in this novel approach, parkinsonian syndromes that used to belong to distinct categories may become grouped together because of a common molecular defect. The challenging nature of the taxonomy of PD is well illustrated in the reported pathologic pleomorphism in patients carrying LRRK2 mutations (13). Although the usefulness of such a proposed recasting would have to be demonstrated, the idea deserves, at the very least, serious consideration as the issue of diagnostic heterogeneity is a well-recognized hurdle for clinical trials geared toward testing neuroprotective agents for PD.
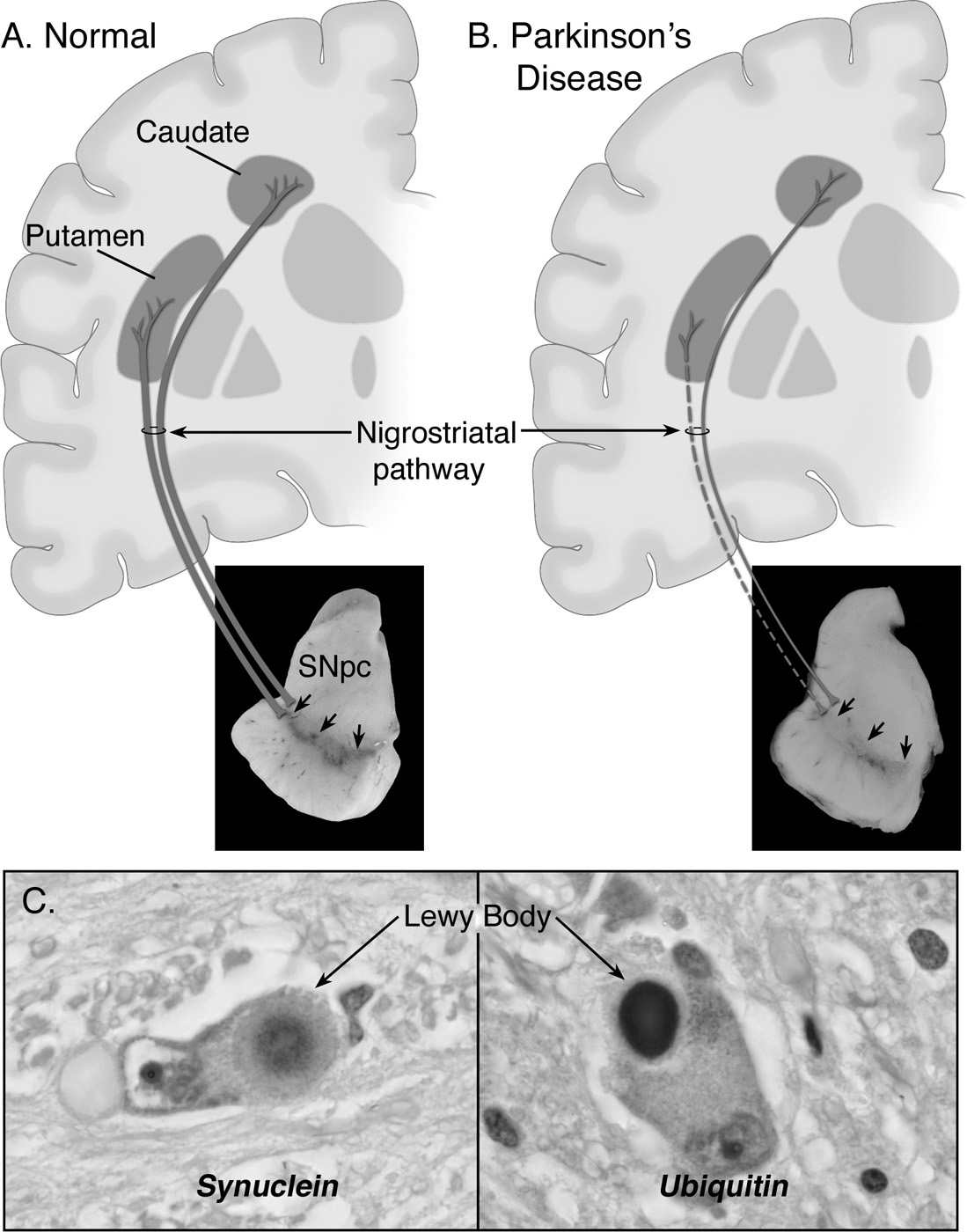
Figure 7.1. Neuropathology of Parkinson’s disease (PD). A: Schematic representation of the normal nigrostriatal pathway (in dark gray). It is composed of dopaminergic neurons whose cell bodies are located in the substantia nigra pars compacta (SNpc; see arrows). These neurons project (thick solid gray lines) to the basal ganglia and synapse in the striatum (i.e., putamen and caudate nucleus). The photograph demonstrates the normal pigmentation of the SNpc produced by neuromelanin within the dopaminergic neurons. B: Schematic representation of the diseased nigrostriatal pathway (in dark gray). In PD, the nigrostriatal pathway degenerates. There is a marked loss of dopaminergic neurons that project to the putamen (dashed line) and a much more modest loss of those that project to the caudate (thin gray solid line). The photograph demonstrates depigmentation (i.e., loss of dark pigment neuromelanin; arrows) of the SNpc due to the marked loss of dopaminergic neurons. C: Immunohistochemical labeling of intraneuronal inclusions (Lewy bodies) in an SNpc dopaminergic neuron. Immunostaining with an antibody against α-synuclein reveals a Lewy body (black arrow) with an intensely immunoreactive central zone surrounded by a faintly immunoreactive peripheral zone (left photograph). Conversely, immunostaining with an antibody against ubiquitin yields more diffuse immunoreactivity within the Lewy body (right photograph). (From Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39:889–909.)
| Parkinsonian Syndromes |
Primary Parkinsonism
Parkinson’s disease (sporadic, familial)
Secondary Parkinsonism
Drug-induced: dopamine antagonists and depletors
Hemiatrophy–hemiparkinsonism
Hydrocephalus: normal-pressure hydrocephalus
Hypoxia
Infectious: postencephalitic
Metabolic: parathyroid dysfunction
Toxin: Mn, CO, MPTP, cyanide
Trauma
Tumor
Vascular: multi-infarct state
Parkinson-Plus Syndromes
Cortical–basal ganglionic degeneration
Dementia syndromes: Alzheimer’s disease, diffuse Lewy body disease, frontotemporal dementia
Lytico–Bodig (Guamanian Parkinsonism–dementia–ALS)
Multiple system atrophy syndromes: striatonigral degeneration, Shy–Drager syndrome, sporadic OPCA, motor neuron disease–parkinsonism
Progressive pallidal atrophy
Progressive supranuclear palsy
Familial Neurodegenerative Diseases
Hallervorden–Spatz disease
Huntington’s disease
Lubag (X-linked dystonia–parkinsonism)
Mitochondrial cytopathies with striatal necrosis
Neuroacanthocytosis
Wilson’s disease
ALS, amyotrophic lateral sclerosis; CO, carbon monoxide; Mn, manganese; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; OPCA, olivopontocerebellar degeneration.
Source: From Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39:889–909.
ETIOLOGIC THEORIES OF SPORADIC PD
The etiology of almost all occurrences of PD remains unknown. In more than 90% of the cases, PD arises as a sporadic condition, that is, in absence of any family history but, in the remaining instances, the disease is unquestionably inherited (Table 7.2). Nonetheless, first-degree relatives of sporadic PD patients are two to three times more likely to have PD than relatives of controls (14). It is therefore not surprising that, until now, all of the hypotheses regarding the etiology of sporadic PD have focused primarily on environmental toxins and genetic factors. As pointed out by Tanner et al. in the previous edition of this chapter (15), the precept about the etiology of PD, for a long time, has been that a single or, at most, a few genes or environmental factors are sufficient to cause PD in most individuals and that both genetic and environmental risks would operate independently. The current school of thought, however, proposes a more multifactorial view of the problem by which diseases, such as PD, would result from a complex interplay of both genetic and environmental factors. Based on this scenario, even in the presence of a known single-gene pathogenic mutation, PD would arise only when both the genetic variant and the deleterious environmental exposure coincide. In this context, a genetic variation would not necessarily cause the disease but would rather influence a person’s susceptibility to environmental factors. The reports of heterozygous mutations in the GBA gene increasing the risk of developing PD by 5 to 6 times (16) has become probably the most prototypical example in support of such a view. Hence, a person may not inherit the disease state per se but rather a set of susceptibility traits to certain environmental factors that place this person at a higher risk of developing PD.
ENVIRONMENTAL TOXINS
According to the environmental hypothesis, PD-related neurodegeneration is provoked by exposure to a dopaminergic neurotoxin. The progressive nature of the neurodegeneration seen in PD would either result from a persistent toxic exposure that causes a sustained insult or from a circumscribed toxic exposure that initiates a self-perpetuating cascade of deleterious events. Relevant to the environmental hypothesis are numerous human epidemiologic studies that have implicated consumption of well water, residence in a rural setting, farming and its associated exposure to herbicides and pesticides with an elevated risk for PD. These epidemiologic notions have been reviewed comprehensively by Tanner et al. in the previous edition of this chapter (15), and will thus not be rehashed. Instead, only selected and recent epidemiology data will be discussed in the following pages.
From the outset, it is stressed that none of the epidemiologic studies to date have convincingly linked a specific environmental toxin to the cause of sporadic PD. Nevertheless, cigarette smoking and coffee drinking are inversely associated with PD (17), reinforcing the concept that environmental factors may indeed contribute to PD susceptibility or etiology. It is thus plausible that the lack of compelling evidence incriminating specific environmental factors simply reflects the limitations of our current analytic methodologies. Moreover, there are several known parkinsonian neurotoxins that potentially could be accumulating in our environment. For instance, exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a by-product of 1-methyl-4-phenyl-4-propionoxypiperidine synthesis (18), causes a clinical condition in humans (19) and several other mammalian species almost identical to PD. Although autopsy studies of MPTP-intoxicated individuals with parkinsonism have consistently demonstrated a profound degeneration of the nigrostriatal dopaminergic pathway, no Lewy body has ever been found in these postmortem brain samples (20,21). Moreover, despite the impressive resemblance between PD and MPTP intoxication, MPTP has never been recovered from brain tissues or body fluids of PD patients.
Another potential environmental toxin is 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of MPTP, that was developed as Cyperquat™, a herbicide (never commercialized), and paraquat, both of which are structurally similar (Fig. 7.2). Both MPP+ and paraquat are polar molecules and, consequently, do not passively cross the blood–brain barrier (BBB). Of these two toxins, only with paraquat has an active transport system been identified at the level of the BBB (22), making it possible, at least, for the latter to accumulate in the brain and inflict damage. Consistent with this view is the demonstration that the systemic administration of paraquat to rodents is associated with the degeneration of nigrostriatal neurons and the formation of Lewy body–like inclusions (23). In a case–control study in Taiwan, chronic exposure to paraquat has also been associated with an increased risk of developing PD (24). Interestingly, as noted by Thiruchelvam et al. (25), manganese ethylene-bis-dithiocarbamate shows a striking geographic overlap with paraquat and has been implicated in cases of PD-like syndromes in agricultural workers. This compound also seems to exacerbate the dopaminergic neurotoxicity of paraquat. If paraquat was an environmental toxicant implicated in PD, the latter observation suggests that one should probably not think of the contribution of an environmental insult to the cause of PD in terms of a single toxin but rather in terms of a combination of toxic agents such as paraquat, manganese ethylenebisdithiocarbamate, and possibly others.
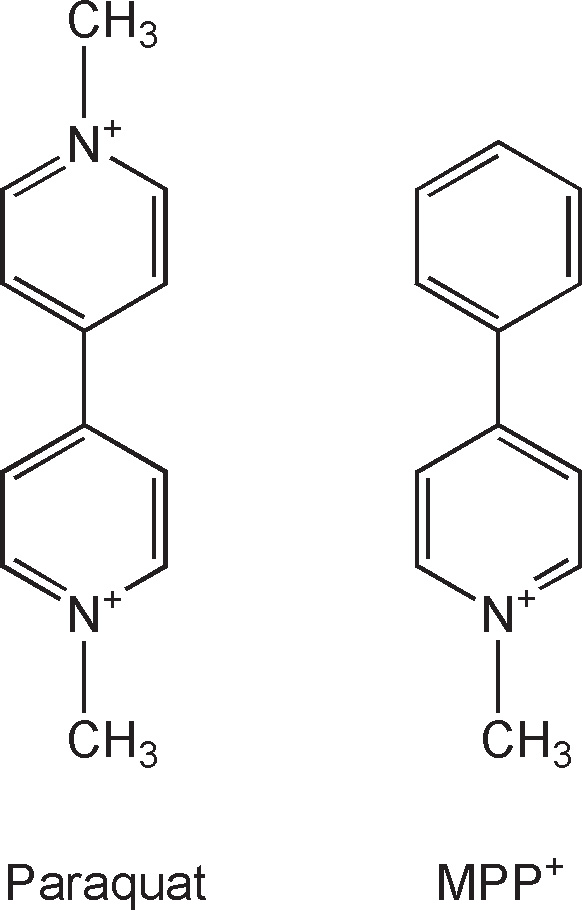
Figure 7.2. Structural similarity between paraquat (left) and MPP+ (right). The only difference between these two compounds is the second N-methyl-pyridium group that paraquat has instead of the phenyl group seen in MPP+.
What about rotenone as an environmental culprit? Rotenone is the most potent member of the rotenoids, a family of natural cytotoxic compounds extracted from various parts of Leguminosae plants. Like MPP+, rotenone is a mitochondrial poison that is present in the environment and abundantly used around the world as an insecticide and a piscicide used to kill unwanted lake fish (26). Although rats chronically intoxicated with rotenone display nigrostriatal damage with Lewy body–like inclusions (27), it is unlikely to cause PD because rotenone is unstable and lasts only a few days in the environment (26). Indeed, rotenone breaks down readily on exposure to sunlight and nearly all of its toxicity is lost in 5 to 6 days of spring sunlight or 2 to 3 days of summer sunlight. Rotenone is also broken down rapidly in soil and in water. Also arguing against an etiologic role for rotenone in PD is the demonstration that the chronic ingestion of rotenone by systemic infusion for 24 months at doses 30 times greater than that used to model PD (27) failed to cause any behavioral or neuropathologic features of the disease in rats (28). Nevertheless, there is one case of fatal rotenone poisoning following its acute ingestion (28). At autopsy, rotenone was found in the blood, liver, and kidney but not in the brain.
Another possibility, which does not, in the strict sense of the word, fall into the environmental category, is that an endogenous toxin may be responsible for PD neurodegeneration. As discussed elsewhere (29), distortions in normal metabolism might create toxic substances because of environmental exposures or inherited differences in metabolic pathways. One obvious source of endogenous toxins may be the metabolism of dopamine, which can generate harmful reactive oxygen species (ROS) (30). Consistent with the endogenous toxin hypothesis is the report that patients harboring specific polymorphisms in the gene encoding for the xenobiotic detoxifying enzyme cytochrome P450 may be at greater risk of developing young-onset PD (31). Further, isoquinoline derivatives and 6-hydroxydopamine, which are both toxic to dopamine neurons, have been recovered respectively from the brain (32) and urine of PD patients (33). Yet the link between polymorphisms in the xenobiotic-metabolizing enzymes and risk of PD remains equivocal, and a comprehensive discussion on this important topic can be found in Paolini (34). From the preceding discussion, it can be concluded that, at this point, the environmental hypothesis of PD remains highly speculative but by no means negligible.
MITOCHONDRIAL GENETICS AND PD
Since the mid-1980s, a mitochondrial defect has been proposed as an etiologic factor in PD, but when one scrutinizes the available evidence, it appears that this appealing notion is nothing more than presumptive. There are at least two main reasons for this continuing appeal. First, as discussed in more detail in Reference 35, it has been noted that, among the large number of genetic loci linked to adult-onset neurodegenerative disorders, including PD, there is a disproportionate number that entertain some form of association with mitochondria. And, second, poisons of the mitochondrial respiratory chain, such as MPTP and rotenone, can give rise to a neurodegenerative phenotype, primarily in animals, reminiscent to that seen in PD (29). Although as discussed below, alterations in mitochondria can be involved in pathologic processes other than oxidative phosphorylation, as for years, the field has been focusing on the hypothesis that PD may be caused by respiratory chain defects in light of a series of descriptive and correlative studies performed on postmortem tissues. Moreover, such a view of PD pathogenesis has ignored the wealth of information originating from patients with true mitochondrial diseases and which should have dampened unjustified enthusiasm.
For instance, real mitochondrial diseases are typically pediatric conditions with a life span expectancy below the age of 40. All mitochondrial DNA of a zygote originate from the ovum, thus it is the traditional view that diseases linked to mitochondrial mutations are maternally inherited. To date, it is true that some epidemiologic evidence supports a maternal inheritance pattern in a subset of PD patients (36,37). Furthermore, polymorphisms in subunits of complex I of the electron transport chain have been proposed as susceptibility genes in subgroups of PD patients (38,39). Although, thus far, true cases of PD linked to a mitochondrial mutation are still lacking, it should be mentioned that a point mutation (A1555G) in the 12SrRNA gene has been implicated in a maternally inherited deafness associated with levodopa-responsive parkinsonism (40). Subsequently, a distinct heteroplasmic, maternally inherited 12SrRNA point mutation (T1095C) was found in another pedigree with deafness, levodopa-responsive parkinsonism, and neuropathy (41). These mutations were not found, however, in 20 cases of sporadic PD, suggesting that the 12SrRNA mutations are not likely to be a common cause of PD. Clearly, when parkinsonism is attributed to one of these rare mitochondrial DNA mutations, it is part of a multisystem clinical picture and not a pure PD presentation. It can thus be concluded that current data supporting the mitochondrial genetic hypothesis of PD etiology seems lacking. That said, mitochondrial alterations may still be instrumental in both the etiology and pathogenesis of PD (see the following) through molecular defects other than a mitochondrial mutation.
PD MENDELIAN GENETICS
Since the discovery in 1997 that missense mutations in α-Synuclein cause a rare form of PD, there has been a resurgence of interest in genetic factors that contribute to the etiology of the disease. As reviewed in detail by Trinh and Farrer (42), at this time, mutations in at least two genes, namely α-Synuclein and LRRK2/dardarin, are associated with dominantly inherited forms of PD and, in three genes, namely Parkin/PARK2, PINK1/PARK6, and DJ1/PARK7, with recessively inherited forms of PD. Of note, in addition to their detection in familial PD, mutations in Parkin and LRRK2 were also found in a number of patients without any apparent family history of PD (43,44). If one allows the inclusion of clinical features such as dystonia, supranuclear palsy, and spasticity in the diagnosis of familial PD, then mutations in VPS35 and ATP3A2 should also be mentioned. Moreover, large-scale genetic epidemiologic studies have revealed a single-nucleotide polymorphism in tau as being associated with PD susceptibility (45), and both apolipoprotein E and probably more than one gene on chromosome 1p have been reported to influence age at onset for PD (46,47); and, as mentioned above, heterozygosity for a mutation in the GBA gene may also predispose to PD (48). Finally, studies of noncoding small RNA species (microRNA) have revealed a number of gene dysregulations that are of potential significance to PD (49).
Worth noting is the fact that studies of monozygotic twins have demonstrated a lack of concordance for PD, and this finding has been frequently cited as evidence arguing against a strong genetic contribution to sporadic PD (50). However, the interpretation of twin studies in PD is often complicated by the fact that twins may be clinically discordant for PD for up to 20 years (51). Moreover, a study utilizing 18F-fluorodopa with PET to investigate dopaminergic function in twin pairs clinically discordant for PD found significantly greater concordance for monozygotic but not dizygotic twins (52). These observations emphasize the need to utilize functional imaging in such studies to identify patients with subclinical disease and suggest that the actual contribution of genetic factors to the causes of PD may be more significant than initially thought.
α-SYNUCLEIN MUTATIONS AND OVEREXPRESSION
Five missense mutations (A30P, E46K, H50Q, G51N, and A53T) in the α-synuclein gene have been linked to a dominantly inherited PD (53–57). Clinical and pathologic features typical of PD have been found in patients with any of the three mutations, although some atypical features have also been noted (54,58). Conversely, α-synuclein mutations have not been found in sporadic PD (59). Therefore, the idea that mutant α-synuclein and sporadic PD share common pathogenic mechanisms relies predominantly on the observation that α-synuclein is a major component of Lewy bodies in sporadic PD (60). Transgenic overexpression of mutant α-synuclein in mice or flies has been generally associated with the development of some of the neuropathologic features of PD, such as intraneuronal proteinaceous inclusions (29). These studies, together with the finding that α-synuclein ablation in mice does not lead to neurodegeneration (61,62), support the concept that α-synuclein mutations operate by a toxic gain-of-function mechanism.
Remarkably, multiplication of the gene encoding for wild-type α-synuclein can also cause an autosomal-dominant PD phenotype (63–65). An important finding that has emerged from these studies is that relatively small alterations in α-synuclein expression can have a marked impact on the disease phenotype. In the study by Chartier-Harlin et al. (64), individuals with α-synuclein gene duplication had a mean age at the onset of 48 years with a disease duration of approximately 17 years. Similarly, in the study by Ibáñez and collaborators (65), both patients with the duplication mutation had onsets of the disease at ages 46 and 50. Conversely, in the report by Singleton et al. (63), affected individuals carried four copies of the α-synuclein gene and developed a rapidly progressive (mean duration: 8 years) and young-onset form of PD (mean age: 38 years). It thus seems that the higher the expression level of α-synuclein, the more malignant the PD phenotype. The fact that increased amounts of wild-type α-synuclein can cause PD also advocates for the idea that the cytotoxic function gained by mutant α-synuclein proteins is not a newly acquired property but rather the enhancement of a native property. However, the nature of the gained function is still enigmatic, in part, because the normal function of α-synuclein is just beginning to be elucidated. The fact that this prevalent presynaptic protein is abundant in Lewy bodies prompted many investigators to believe that its propensity to misfold and form α-synuclein fibrils may be responsible for its neurotoxicity. More can be read about the hypothesized toxic mechanisms of α-synuclein references 29 and 66.
PARKIN MUTATIONS
Loss-of-function mutations in the gene encoding Parkin cause a recessive-inherited parkinsonism (67). Although this form of parkinsonism was originally termed autosomal recessive juvenile parkinsonism, the clinical phenotype includes a man with onset at age 64 and other late-onset patients. In general, however, Parkin mutations are found in PD patients with onset before age 30, particularly those with a family history consistent with recessive inheritance (68). Clinically, Parkin mutant patients display the classic signs of parkinsonism but with marked improvement of symptoms with sleep, abnormal dystonic movements, and a striking response to levodopa. Heterozygote mutations in Parkin may also lead to dopaminergic dysfunction and later onset of parkinsonism, consistent with a mechanism of haploinsufficiency (69,70). Pathologically, parkin-related PD is characterized by the loss of substantia nigra pars compacta (SNpc) dopaminergic neurons, but it is not typically associated with Lewy bodies (68), in that Lewy bodies were definitely identified in only a single patient harboring a parkin mutation (64). Unexpectedly, attempts to recapitulate Parkin loss of function in mice and flies have not succeeded in producing a conclusive PD phenotype (71–73). Nevertheless, these engineered animals are not completely normal, as they appear to exhibit mitochondrial defects (72–74), suggestive of those described in sporadic PD (29).
To date, it is uncertain how Parkin mutations lead to dopaminergic neuron degeneration, but one clue has emerged from the identification of its normal function, which is as an E3 ubiquitin ligase (75,76), a component of the ubiquitin–proteasome system. The way E3 ligases work is by conferring target specificity through their capacity to bind to specific molecules or classes of molecules, thereby facilitating the polyubiquitination necessary for targeting to the proteasome. More recently, it has been found that parkin, primarily a cytosolic protein, can translocate to dysfunctional mitochondria and participate in the destruction of these defective organelles by macroautophagy (77). Also noteworthy is the lack of Lewy bodies in most PD patients carrying parkin null mutations (78), suggesting that parkin E3 ligase activity might be needed for Lewy body formation. At this point, however, what is the link among these different parkin biologic functions and how, given these putative roles, Parkin mutations cause PD remain enigmatic.
DJ-1 MUTATIONS
DJ-1 is a homodimeric, multifunctional protein ubiquitously expressed in human tissues, including the brain. Thus far, at least 11 different DJ-1 mutations, including missense, truncating, splice-site mutations and large deletions, have been linked to an autosomal recessive form of PD (79–81). Although DJ-1 mutations account for a small fraction of familial PD, it is now recommended to systematically screen for DJ-1 mutations in all cases of recessively inherited early-onset forms of the disease (82). These mutations are found throughout four of the seven exons of the DJ-1 gene. Although little is known about the function of DJ-1, it is predicted that many, if not all of the identified mutations, would cause either a lack of DJ-1 expression or the transcription of an unstable and functionally deficient product.
Cells lacking DJ-1 have been reported to be more susceptible to a variety of stresses, including oxidative attack. These findings prompted researchers to propose that DJ-1 protects cells through some kind of antioxidant effect. However, whether this is indeed the case is still a matter of intense investigation. Structural studies also indicate that DJ-1 shares similarities with the bacterial heat shock protein (Hsp)-31 a stress-inducible chaperone (83). In keeping with this observation is the demonstration that wild-type DJ-1 does exhibit chaperone activity (84). However, this Hsp-31-like activity is not detectable when DJ-1 is in reducing conditions (which mimics the physiologic intracellular environment), but is detectable when in oxidative conditions (which mimics an oxidative stress situation) (84). Furthermore, the PD-associated DJ-1 mutation L166M abolishes this redox-dependent chaperone activity (84). Collectively, these data are consistent with the emerging idea that wild-type DJ-1 functions as a redox-dependent chaperone that becomes activated to assist the cell in coping with the rising amount of oxidatively damaged, misfolded proteins in response to intracellular oxidative stress.
PINK-1 MUTATIONS
Autozygosity mapping of a large consanguineous Sicilian family located the PARK6 locus linked to an autosomal recessive form of PD to chromosome 1p35–36 (85). PARK6 was then linked to an early-onset recessive PD in eight additional families from four different European countries (86
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


