INTRODUCTION
Gliomas are a group of primary central nervous system (CNS) neuroepithelial tumors that appear to arise from glial progenitor cells in the brain or spinal cord. Based on their resemblance to glial lineages, gliomas have been classified into different tumor types, including astrocytomas, oligodendrogliomas, mixed oligoastrocytomas, ependymomas, and several less common types of glioneuronal tumors. Gliomas are the most common type of tumor arising in the brain. Because they rarely metastasize outside of the CNS, gliomas are not staged like systemic malignancies. Instead, gliomas are graded based on histopathologic features that predict aggressiveness; tumors with malignant features are often referred to as high-grade gliomas (HGGs), in contrast to “low-grade” gliomas (LGGs), which in spite of their more benign histologic appearance, are not clinically benign. The location and invasiveness of most LGGs that occur in adults often makes them difficult to completely resect, leading to recurrent disease, as well as significant morbidity and mortality. The lowest grade gliomas (such as pilocytic astrocytomas) are an exception and are often surgically curable but they rarely occur in adults.
EPIDEMIOLOGY
Gliomas are relatively rare tumors with an estimated annual incidence of 4.67 to 6.02 per 100,000 persons. HGGs have an increasing incidence with age and are significantly more common than their low-grade counterparts, which appear more frequently in young adults. In general, the incidence of gliomas is higher in men than in women and in non-Hispanic whites than in blacks, Hispanics, Asians, or Native Americans.
There are very few known risk factors for the development of glioma, and no preventative measures or lifestyle changes are known to decrease an individual’s risk. Several rare genetic tumor predisposition syndromes are associated with increased glioma incidence, including the neurofibromatoses, tuberous sclerosis, Cowden, Li-Fraumeni, and Turcot syndromes. Exposure to ionizing radiation is the only established environmental risk factor that can lead to glioma development. Frequent concerns about head injury, pesticides, occupational exposures, and foods containing N-nitroso compounds or aspartame appear to be unfounded or at least unproven. Although there has been concern about cell phones and other electromagnetic fields, existing evidence is conflicting and often limited by confounding recall bias and other shortcomings of retrospective study design. To date, there is no convincing evidence that cell phone use increases the risk of brain tumors, although studies continue.
CLINICAL FEATURES
Like patients with brain metastases, patients with gliomas present with diverse nonspecific clinical symptoms and signs that are related to tumor location, size, and growth rate rather than tumor type, except perhaps seizures which may be more common from oligodendrogliomas. Focal symptoms such as hemiparesis, aphasia, ataxia, visual field deficits, or cranial neuropathies result from localized invasion or compression of critical brain structures, whereas generalized symptoms such as headache, vomiting, lethargy, and confusion tend to be the result of increased intracranial pressure from tumor mass, edema, or hydrocephalus. Seizures resulting from an underlying glioma invariably begin focally but may rapidly generalize, making it difficult to recognize their partial onset. Although seizures or symptoms from hemorrhage appear suddenly, most other symptoms produced by gliomas present subacutely over days to weeks. HGGs tend to present with more rapidly progressive focal or generalized symptoms, whereas LGGs frequently precipitate seizures without progressive neurologic deficits. Depending on location, even large tumors can produce surprisingly few symptoms, as in the case of LGGs, to which the brain can gradually adapt. Consequently, an apparently normal neurologic exam cannot rule out an underlying glioma, and a thorough history and physical exam must be coupled with a high degree of clinical suspicion in order to know when to consider the diagnosis and obtain neuroimaging.
PATHOBIOLOGY
The diagnosis of a glioma is made based on histopathologic appearance under light microscopy, occasionally aided by immunohistochemistry and specific molecular testing. Morphologic similarity to normal glial cells is used to assign tumors to particular glioma subtypes, including astrocytoma, oligodendroglioma, mixed oligoastrocytoma, ependymoma, and glioneuronal tumors. Within each of these tumor types, the presence or absence of specific anaplastic features is used to infer tumor behavior and growth rate and to assign tumors to pathologic grades, with implications for prognosis and treatment. Some of the features that determine tumor grade include cellular atypia, anaplasia, mitotic activity, endothelial proliferation, and necrosis. Gliomas are graded using the World Health Organization (WHO) classification system that includes grades I to IV. WHO grades I and II are considered low-grade tumors, whereas WHO grades III and IV are considered high-grade, malignant tumors. Based on their pathologic appearance, LGGs have occasionally been referred to as benign; however, this term is a misnomer, as the clinical course of LGGs in adults almost always includes recurrence and progression to higher grade malignancy and death (except the lowest grade tumors such as pilocytic astrocytomas described further in the following text).
Glioma grade is determined by the highest grade features identified, even if those features are restricted to a small region within the tumor. Because of the location and marked heterogeneity of many gliomas, there is a risk of sampling error and undergrading, particularly when the diagnosis is made based on a stereotactic biopsy.
A rapidly increasing understanding of the molecular pathogenesis of gliomas has the potential to improve diagnosis and lead to the discovery of more effective treatments. Currently, there are a number of molecular biomarkers used to assist in establishing the diagnosis, estimating prognosis, and predicting treatment responsiveness in several glioma subtypes. Numerous additional molecular features are being studied as markers or potential targets of molecularly directed therapeutics in an attempt to improve glioma treatment.
ASTROCYTOMAS
PILOCYTIC ASTROCYTOMA
Definition and Epidemiology
Pilocytic astrocytomas are relatively well-circumscribed World Health Organization (WHO) grade I astrocytic gliomas that primarily occur in children and young adults. Pilocytic astrocytomas make up 5% to 6% of all gliomas and are the most common glioma in children but only rarely appear in older adults. They exhibit slow indolent growth, and unlike most other LGGs, can often be cured with complete resection. Pilocytic astrocytomas are associated with neurofibromatosis type I where they are estimated to occur in 15% to 20% of patients.
Clinical and Imaging Features
Pilocytic astrocytomas can occur anywhere in the neuraxis, although they most frequently arise in the cerebellum. They also have a predilection for the anterior optic pathway, particularly in NF1, where they display an even more indolent course than sporadic tumors. In fact, optic pathway pilocytic astrocytomas are so characteristic of NF1 that any child diagnosed with this tumor should be evaluated for NF1. Pilocytic astrocytomas present insidiously with symptoms related to their location. Infratentorial tumors often present with progressive headache, nausea, and vomiting related to hydrocephalus, whereas optic pathway tumors present with progressive visual loss or proptosis. Seizures are an uncommon presentation, as these tumors rarely involve cerebral cortex. On MRI, pilocytic astrocytomas appear wellcircumscribed with bright contrast enhancement and frequent cyst formation. Despite their defined border, pilocytic astrocytomas do not have a true capsule, and although they are considered noninvasive, they often microscopically infiltrate surrounding brain parenchyma.
Pilocytic astrocytomas generally maintain their WHO grade I status for years and only rarely undergo malignant transformation. Approximately 60% to 80% of sporadic non-NF1-associated pilocytic astrocytomas have been shown to contain a novel BRAF-KIAA
fusion that appears to predict improved survival following partial resection and raises the possibility that these tumors could be treated with BRAF inhibition.
DIFFUSE ASTROCYTOMA
Definition and Epidemiology
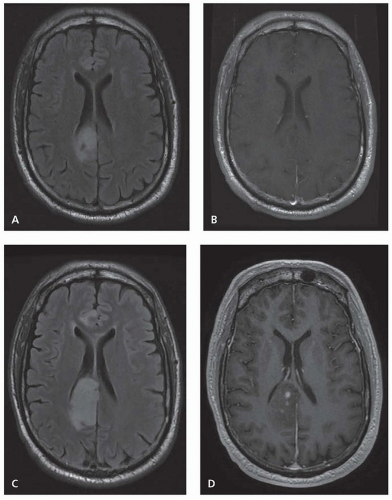
Diffuse astrocytomas are WHO grade II infiltrative astrocytic tumors that typically affect otherwise healthy young adults. They are fairly well-differentiated tumors with a slow, indolent early course, although they have the tendency to eventually undergo malignant transformation to high grade (
Fig. 96.1). Diffuse astrocytomas make up 10% to 15% of all astrocytic tumors with an annual incidence of 1.4 per 1 million and a peak incidence in the fourth decade of life.
Clinical and Imaging Features
Diffuse astrocytomas can occur anywhere in the CNS, although they preferentially arise in the cerebral hemispheres in adults. In children, they often occur in the brain stem as diffuse intrinsic pontine gliomas. Diffuse astrocytomas most frequently present with seizures but depending on location can also cause focal symptoms or cognitive and behavioral change. Diffuse astrocytomas generally appear as ill-defined homogeneous regions with T1 hypointense signal and T2/FLAIR hyperintensity, although they tend to infiltrate beyond the T2/FLAIR margins visible on MRI. Diffuse astrocytomas generally have minimal contrast enhancement, and the
development of new contrast enhancement can signal progression to higher grade (see
Fig. 96.1).
Pathologic and Molecular Features
Diffuse astrocytomas are made up of well-differentiated astrocytes with increased cellularity and occasionally demonstrate nuclear atypia but rare mitotic activity, with Ki-67/MIB-1 staining growth fraction typically less than 4%. By definition, diffuse astrocytomas have no necrosis or endovascular proliferation. Although they appear fairly benign histologically, diffuse astrocytomas are not clinically benign, with a high rate of transformation to malignancy over the course of years. Point mutations in the isocitrate dehydrogenase enzyme IDH1 (R132), or rarely IDH2 (R172), are present in the majority (65% to 80%) of grades II and III gliomas, including 75% of diffuse astrocytoma. The presence of an IDH mutation is a favorable prognostic factor regardless of tumor type, and IDH mutation can be detected by immunohistochemistry, polymerase chain reaction (PCR), and MR spectroscopy. Methylation of the O6-methylguanine-DNA methyltransferase (MGMT) promoter, which leads to MGMT gene silencing is also common in diffuse astrocytoma and is associated with improved prognosis and alkylating agent chemosensitivity.