Fig. 9.1
Cutaneous manifestations of necrolytic migratory erythema. Glossitis (a) may be associated with the cutaneous lesions (b). The lesions are erythematous and polycyclic. The skin biopsy (c) shows a parakeratotic psoriasiform epidermis with superficial cleft, necrolysis in the upper epidermis, vacuolated keratinocytes, and mild perivascular lymphocytic infiltration
The typical feature in skin biopsy is necrolysis of the upper epidermis with vacuolated keratinocytes (Fig. 9.1c). Psoriasiform epidermal hyperplasia, hypogranulosis, and thickened stratum corneum with parakeratosis as well as perivascular lymphohistiocytic infiltrate in the superficial dermis are characteristic histopathologic features [11, 14]. Similar lesions of the skin can occur in pseudoglucagonomas without pancreatic tumor, usually due to generalized malabsorption syndrome or specific nutritional deficiencies (i.e., zinc and essential fatty acids). Same aspects on skin biopsy can be found in other deficiency dermatoses, such as pellagra or acrodermatitis enteropathica [15]. Necrolytic migratory erythema may be misinterpreted as other dermatoses (pemphigoid, mucocutaneous candidiasis, psoriasis, eczema, seborrheic or contact dermatitis) [15]. Due to nonspecific aspects of the cutaneous lesions and the rarity of this kind of tumor, skin involvement precedes the diagnosis of glucagonoma of 6–8 years on average [13].
Worsening or onset of diabetes mellitus is frequent (70–87 %) due to the plasmatic secretion of glucagon [9, 13]. It can be a glucose intolerance or a diabetes necessitating oral antidiabetics or insulin and can be cured with glucagon normalization. Weight loss is usually significant, higher than 20 kg and very evocative of the disease because, in general, it is not present in case of well-differentiated, slow-growing NET. It is secondary to the hypercatabolism of glucagon and is not a prognostic factor [13]. Venous thromboembolisms occur sometimes (25 %), and few cases of dilated heart disease with improvement of the cardiac function with the decrease of the plasmatic glucagon are described [16]. Diarrhea can be present (14 %) without malabsorption but conversely with villous hypertrophy. Finally, psychiatric disorders or deficiency encephalopathies have been observed [13].
Biologically, except diabetes, normochromic and normocytic anemia is characteristic and is related to the severity of the glucagonoma syndrome [9, 17]. Hypoaminoacidemia can occur in 40 % of the glucagonoma syndrome with frequent hypoalbuminemia [9, 13].
When the clinical diagnosis of glucagonoma syndrome is suggested, it should be confirmed by the dosage of the plasma glucagon. The excessive secretion is usually obvious [9, 13]. In basal conditions, plasma glucagon is continuously increased (N, 150 pg/ml), very often greater than 1,000 pg/ml. Hyperglucagonemia less than 500 pg/ml can be observed in other pathological conditions: diabetes mellitus (except those following pancreatectomy or pancreatitis), insufficiency of glucagon catabolism (renal failure, severe liver disease), serious diseases with hypercatabolism (infections, stress, burns), and hyperglucagonemia family. However, usually, differential diagnoses in these conditions are not a problem [13, 17].
After the biological confirmation of the glucagonoma syndrome, morphological exams have to be done to localize the primary tumor, always located in the pancreas [8, 9], more often (53 %) in the tail [7, 9]. At diagnosis, the primary is usually large, greater than 3 cm in 60 % of cases and greater than 10 cm in 20 % of cases according to Guillausseau [5]. The disease is often malignant at diagnosis, mostly (80 %) with liver metastases [7–9]. For these reasons, CT scan with arterial and venous phases (Fig. 9.2a) or MRI (Fig. 9.2b) has a good diagnostic accuracy and shows a hypervascularized tumor without necessity of EUS as in smaller tumor (insulinoma or gastrinoma). Due to the presence of somatostatin receptors, somatostatin receptor scintigraphy is usually positive and can be helpful for the confirmation of the neuroendocrine origin and the staging (Fig. 9.2c, d).
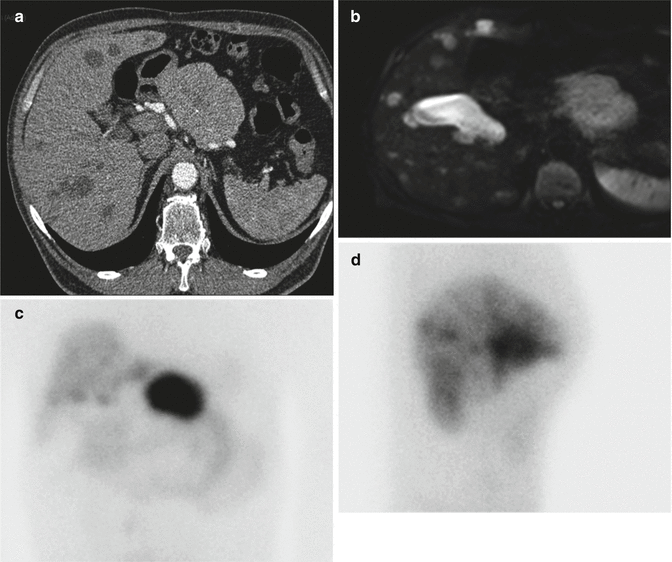
Fig. 9.2
Imaging of a pancreatic glucagonoma with liver metastasis. CT scan (a) shows a large tumor on the left of the pancreas without enhancement on the arterial phase and hypodense bilobar liver metastases. MRI (b) shows same lesions with very hyperintense liver metastases on T2 phase due to their cystic appearance which can explain a slight uptake on the Octreoscan ® of the liver in contrast to the important uptake of the pancreatic primary (c, d)
9.4 Morphology
9.4.1 Macroscopy
Glucagonomas are pancreatic, usually single large tumors. Their diameter is mainly comprised between 3 and 7 cm, but they vary in size from 2 to 25 cm. They frequently occur in the tail of the pancreas [18]. Like most other PanNETs, they are well demarcated from the adjacent pancreas, but in some cases, they can infiltrate the parenchyma (Fig. 9.3a, b). They can be cystic [19].
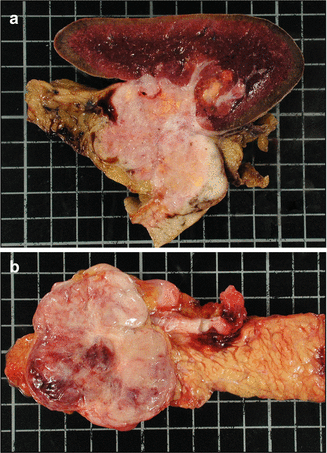
Fig. 9.3
Macroscopic appearance of glucagonomas. Two large, 6 cm-diameter glucagonomas of the caudal pancreas in two different patients, infiltrating the pancreas, peripancreatic adipose tissue, and spleen (a) or better limited (b). These tumors contain small areas of necrosis and hemorrhage
9.4.2 Histology
The histological features of glucagonomas are quite the same as those of other well-differentiated PanNETs (Fig. 9.4a). They show a predominance of a mixed trabecular-solid patterns [10]. No poorly differentiated functioning glucagonoma has been described.
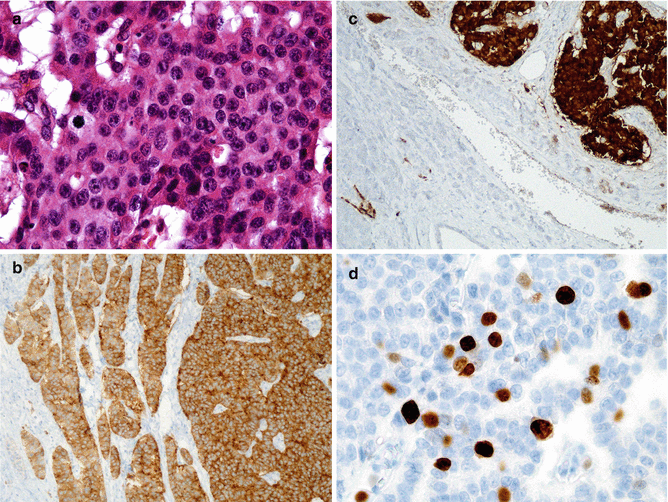
Fig. 9.4
Histologic appearance of glucagonomas. This glucagonoma is a well-differentiated neuroendocrine tumor, grade 2. Some mitoses are present. Tumor cells are regular, and anisonucleosis is mild (a; H&E staining × 40). Tumor cells diffusely express synaptophysin (b; immunohistochemistry × 20). Tumor cells (top right) strongly express glucagon; some cells are detected in adjacent islets in the surrounding parenchyma (c; immunohistochemistry × 20). Ki67 is expressed by 8 % of tumor cells in this grade 2 tumor according to WHO 2010 classification (d; immunohistochemistry; ×40)
9.4.3 Immunohistochemistry
As other PanNETs, glucagonomas express both synaptophysin (Fig. 9.4b) and chromogranin. They stain for glucagon (Fig. 9.4c). PP-positive cells can also be frequently identified in glucagonomas. Glucagonomas can also produce peptides derived from proglucagon (glycentin, glucagon-like peptides 1 and 2) [10].
PanNET can produce more than one type of hormone, including glucagon, especially in the context of MEN1 disease [20, 21]. Presence of cells expressing glucagon can be observed in non-glucagonoma PanNETs. In MEN1, the most frequent tumors are mainly glucagon positive, followed by PP, insulin, and somatostatin. However, glucagon-positive NETs in the context of MEN1 are usually nonfunctioning [20]. The glucagonoma syndrome may be associated with or followed by another syndrome (insulinoma, VIPoma syndromes, etc.). A secretion of parathyroid hormone-related peptide (PTHrP) was described in a patient presenting a large pancreatic glucagonoma with hypercalcemia [22].
9.4.4 Grading and Predictive Factors
There are few data on grading in the literature (Fig. 9.4d). The retrospective series from Wermers et al. describing 21 cases in 1996 and from Vauleon in 2004 do not give any data on proliferation [12, 18]. In Eldor’s study, the Ki67 was available in only 2/6 cases, evaluated at 2 % and 4 % [23].
Related posts:
 Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
The Role of Nuclear Medicine in the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Immunohistochemical Approach to the Diagnosis and Prognostic Evaluation of Pancreatic Neuroendocrine Neoplasms
Mixed Adenoneuroendocrine Carcinoma of the Pancreas
ACTH-Producing Tumor
Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
The Role of Nuclear Medicine in the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Immunohistochemical Approach to the Diagnosis and Prognostic Evaluation of Pancreatic Neuroendocrine Neoplasms
Mixed Adenoneuroendocrine Carcinoma of the Pancreas
ACTH-Producing Tumor
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


