Huntington Disease
Karen S. Marder
INTRODUCTION
Huntington disease (HD) is an autosomal dominant neurodegenerative disorder caused by a cytosine-adenine-guanine (CAG) polyglutamine repeat expansion in exon 1 of the Huntington (HTT) gene on chromosome 4p16.3. The age of onset, defined based on the presence of a characteristic extrapyramidal movement disorder, is inversely related to CAG repeat length such that higher CAG repeat length is associated with earlier age at onset. HD is characterized by a triad of symptoms and signs including motor, cognitive, and psychiatric manifestations. Studies of premanifest HD have demonstrated changes in imaging biomarkers (putamen and caudate) that predate onset of motor signs more than 15 years prior to onset and cognitive changes that occur 10 years prior to motor onset. Symptomatic treatment for chorea but no diseasemodifying therapy is currently available. The development of imaging, biofluid, and quantitative cognitive and motor testing will facilitate clinical trial development in prodromal HD.
EPIDEMIOLOGY
James Huntington provided the first comprehensive description of the disease and its inheritance pattern in 1872. Since that time, it has been described worldwide. Prevalence of HD based on a meta-analysis of 13 “service-based” studies was 2.71 per 100,000 (95% confidence interval [CI], 1.55 to 4.72). Prevalence was significantly lower in Asia 0.4 per 100,000 compared to Europe, North America, and Australia 5.7 per 100,000. Among people of European ancestry, 4 to 7 per 100,000 people have HD, with far lower prevalence among Asians and Black Africans. Incidence based on four studies was 0.38 per 100,000 per year (95% CI: 0.16, 0.94), with incidence much lower in Asia compared to those of European ancestry. Seven common haplotypes have been identified in the HTT gene, with a single ancestral haplotype accounting for 50% of all European chromosomes, supporting the theory that HD may have originated in Northern Europe and spread worldwide.
PATHOBIOLOGY
GENETICS
Age of onset of HD, defined by the presence of extrapyramidal motor signs, occurs most often between 40 and 50 years (mean age 45 years); however, cases have been reported as young as age 2 years and as old as 85 years. Between 5% and 10% develop HD before age 20 years (juvenile HD) and about 10% after age 60 years. CAG repeat length is inversely correlated with age at onset and accounts for 50% to 70% of the variance in age at onset. Repeat lengths of 26 or less are normal. Repeats between 27 and 35 are considered intermediate or “high normal” and represent 3% to 5% of all “normal” alleles. Although carriers of 27 to 35 repeats will not develop manifest HD, this range confers meiotic instability, and parents, especially fathers, may transmit repeats to their children that are within the HD range. Carriers of 27 to 35 repeats may evidence subtle behavioral changes, an “endophenotype,” although they do not have motor or cognitive impairment. Repeats between 36 and 39 are associated with incomplete or reduced penetrance, meaning that some individuals will develop HD, generally late onset, and others will not. Diagnosis of HD in the 36 to 39 repeat range may be difficult due to confounding with ageassociated signs. Individuals with 40 or more repeats will develop HD (fully penetrant) if they live long enough. Most individuals with HD have between 40 and 50 repeats. Those with juvenile HD usually have greater than 50 repeats, although greater than 100 repeats have been reported.
There is a greater chance for CAG expansion in sperm than in ova, which explains the concept of “genetic anticipation,” an earlier age at onset in the next generation. Mothers with HD pass on approximately the same number of repeats (±3), whereas fathers with HD, because of meiotic instability, often pass on a higher CAG repeat length. Ninety percent of juvenile HD cases have inherited the disease from their fathers. Despite the inverse correlation with age, there is significant variability of age of onset of HD even among individuals with the same CAG repeat length.
NEUROPATHOLOGY
Neuronal loss begins in the striatum. A five-point grading system based on macroscopic and microscopic criteria correlates with the Shoulson-Fahn Total Functional Capacity scale (Fig. 82.1). The scale ranges from 0 (no pathology) to grade 4. The earliest changes are seen in the medial paraventricular portions of the caudate nucleus (50% of neurons are lost in grade 1), in the tail of the caudate, and in the dorsal putamen. Ninety-five percent of neurons are lost in grade 4. Astrocytes increase steadily from grade 2 to 4, with severe gliosis in stage IV. Although neuronal loss in the striatum is the hallmark of HD, extrastriatal involvement (globus pallidus, subthalamic nucleus, substantia nigra pars reticulata, thalamus, brain stem, and neocortex) occurs but is less severe than the striatum (see Fig. 82.1).
Microscopically, the caudate contains shrunken neurons and ubiquitinated, round intranuclear inclusions. These inclusions consist of large aggregates of abnormal huntingtin protein (HTT) (Fig. 82.2B). The normal function of HTT, a predominantly cytoplasmic protein, is not completely understood. HD is believed to arise primarily from a gain of toxic function from an abnormal conformation of mutant HTT; however, loss of function of HTT and toxicity from RNA may also contribute. HTT is essential for early embryonic development. Despite widespread expression of abnormal HTT, the striatum is relatively selectively vulnerable. Subcortical prefrontal white matter shows CD68-labeled microglial cells and macrophages, which probably reflects the slow ongoing widespread degenerative process involving the cortical neurons and their processes (Fig. 82.2A).
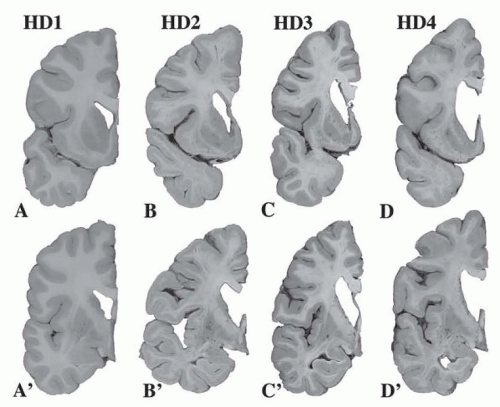 FIGURE 82.1 Gradations of Huntington disease pathology. Neuropathologic gradations of Huntington disease using a 5-point scale (0 to 4). A-D: Coronal sections passing through the nucleus accumbens including the head of the caudate nucleus and putamen. A’-D’: Coronal sections passing through the globus pallidus including the putamen and caudal segment of the head of the caudate nucleus. A and A’: Neuropathologic grade of severity 1 (HD1): (about 5% of all HD brains) there is minimal detectable change on gross examination at the sites shown in A and A’; however, the body and tail of the caudate nucleus are atrophic. B and B’: Neuropathologic grade of severity 2 (HD2): (about 15% of HD brains) the volume loss of the neostriatum is moderate with relative preservation of its shape. The medial outline of the head of the caudate nucleus is only slightly convex, but it still bulges into the lateral ventricle (B). The medial edge of the lenticular nucleus (B’) lost its medial convexity. C and C’: Neuropathologic grade of severity 3 (HD3): (about 50% of HD brains) the medial outline of the head of the caudate nucleus forms a straight line or is slightly concave medially (C). The volume loss of the lenticular nucleus (putamen and globus pallidus) is discrete, and its medial outline is parallel to the anterior limb of internal capsule or is slightly concave medially (C′). D and D’: Neuropathologic grade of severity 4 (HD4): (about 30% of HD brains) the striatum is severely atrophic. The medial contour of the head of the caudate nucleus is concave, as is the anterior limb of internal capsule (D). (Courtesy of JP Vonsattel, MD.) |
NEUROCHEMISTRY
Up to 95% of the GABAergic medium spiny neurons that project to the globus pallidus and the substantia nigra are lost, whereas large cholinergic interneurons are selectively spared. Loss of the medium spiny neurons may be due to excessive stimulation of excitatory amino acid receptors, especially N-methyl-D-aspartate (NMDA) receptors. Although dopamine, glutamate, and γ-aminobutyric acid (GABA) are thought to be most affected in HD and have been targets for therapy, multiple neurotransmitters and their receptors are involved. Cannabinoid and adenosine A2a receptor binding in the striatum and globus pallidus externa are affected early. There is typically loss of dopamine D1 receptors in the striatum as well as cannabinoid and D1 receptors in the substantia nigra as the disease progresses. Loss of substance P/dynorphin neurons occurs in the late stages and correlates clinically with dystonia.
CLINICAL MANIFESTATIONS
The triad of symptoms and signs associated with HD is an extrapyramidal movement disorder, cognitive impairment, and behavioral impairment (Table 82.1). Any one of the triad may occur first. Three carefully designed observational studies including individuals at risk for HD and early symptomatic HD (PHAROS, PREDICT HD, TRACK HD) have facilitated in-depth characterization of individuals followed longitudinally including the period of phenoconversion. The course of HD can be divided into several periods. During the premanifest period (from birth to approximately 10 to 15 years prior to phenoconversion), individuals are clinically indistinguishable from controls (presymptomatic phase). The prodromal phase, which follows, is characterized by subtle motor, cognitive, and psychiatric features leading up to phenoconversion. Phenoconversion is defined clinically, as unequivocally manifesting the extrapyramidal motor signs of HD, when HD is diagnosed.
MOTOR SIGNS
The extrapyramidal movement disorder is the defining feature of HD and is characterized by both involuntary movements (chorea, dystonia) and impairment in voluntary movements (eye movements, gait, swallowing). The Unified Huntington’s Disease Rating Scale includes a 124-point assessment of the motor signs of HD including eye movements (reduced saccades and breakdown of smooth pursuit), fine motor coordination, chorea, dystonia, and gait and balance (Video 82.1). Chorea, derived from the Greek word “dance,” is an involuntary movement that is brief but not as lightening-like as myoclonus. These random movements may be present in the limbs, face, or trunk (see Video 82.1). They are exacerbated with stress and are thought to disappear during sleep.
Chorea tends to be present in the early symptomatic phase, peak mid disease, and decline or disappear in the late phase of the disease. In contrast, bradykinesia, dystonia, and rigidity increase, particularly during the late stages of the disease, and affect voluntary movements. Most individuals need some assistance with walking, for example, assistive devices, approximately 7 years from onset of motor signs. Falls and choking are frequent mid- to late-stage events that may be life threatening. In addition to chorea, other motor features include motor impersistence. Examples include the inability to maintain tongue protrusion for 10 seconds or to maintain voluntary contraction while gripping, colloquially known as milkmaid grip. Impairments in fine motor activity that may include slowing and irregularity can be measured clinically or with quantitative motor assessment including speed of finger tapping and paced tapping. Variability of the inter tap interval or the pace of walking are among the earliest detectable signs of motor impairment, at a time when impairment is not detectable on neurologic examination or recognized by the individual. The earlier the disease onset, the less likely there will be chorea. Children presenting with HD present with an akinetic rigid form of the disease. Stiffness in the legs may also be accompanied by scissoring of gait, bradykinesia, and clumsiness. Seizures occur in 25% of juvenile HD cases.
Chorea tends to be present in the early symptomatic phase, peak mid disease, and decline or disappear in the late phase of the disease. In contrast, bradykinesia, dystonia, and rigidity increase, particularly during the late stages of the disease, and affect voluntary movements. Most individuals need some assistance with walking, for example, assistive devices, approximately 7 years from onset of motor signs. Falls and choking are frequent mid- to late-stage events that may be life threatening. In addition to chorea, other motor features include motor impersistence. Examples include the inability to maintain tongue protrusion for 10 seconds or to maintain voluntary contraction while gripping, colloquially known as milkmaid grip. Impairments in fine motor activity that may include slowing and irregularity can be measured clinically or with quantitative motor assessment including speed of finger tapping and paced tapping. Variability of the inter tap interval or the pace of walking are among the earliest detectable signs of motor impairment, at a time when impairment is not detectable on neurologic examination or recognized by the individual. The earlier the disease onset, the less likely there will be chorea. Children presenting with HD present with an akinetic rigid form of the disease. Stiffness in the legs may also be accompanied by scissoring of gait, bradykinesia, and clumsiness. Seizures occur in 25% of juvenile HD cases.
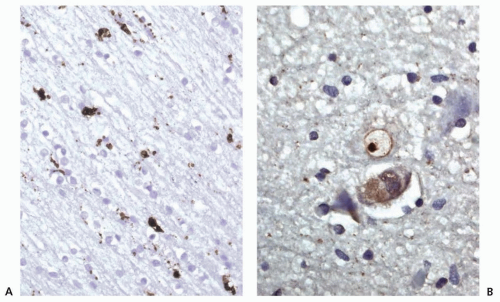 FIGURE 82.2 Neuronal intranuclear inclusion. A: The presence of CD68-labeled microglial cells and macrophages within the subcortical prefrontal white matter (Brodmann area [BA] 9) is shown. B: Two neurons from putamen (HD3-neuropathologic staging) are shown. One of them exhibits a ubiquitinated, round nuclear inclusion. The other neuron is shrunken and diffusely labeled with antibodies directed against ubiquitinated proteins. (Courtesy of JP Vonsattel, MD.) |
TABLE 82.1 Clinical Manifestations of Huntington Disease
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|
