INTRODUCTION
Inherited neuropathies, collectively known as Charcot-Marie-Tooth (CMT) disease, are a group of genetically and phenotypically heterogeneous peripheral neuropathies associated with mutations or copy number variations in over 70 distinct genes. Named after the three neurologists who first described the condition in 1886, CMT is the most common inherited neuromuscular disease. CMT is a motor and sensory neuropathy (HMSN) that is closely related to two other rarer inherited neuropathies: distal hereditary motor neuropathy (dHMN), which has predominantly motor involvement, and hereditary sensory/autonomic neuropathy (HSN or HSAN), which involves only or predominantly sensory and autonomic nerves. These three disorders represent a continuum and are often collectively termed Charcot-Marie-Tooth and related disorders.
CMT is divided into different forms based on the pattern of inheritance and neurophysiologic studies. Autosomal dominant forms are subdivided into demyelinating (CMT1) and axonal (CMT2) forms. CMT4 and CMTX designate the autosomal recessive and X-linked forms, respectively. The term
Dejerine-Sottas neuropathy (DSN) is currently used primarily to denote severe early-onset clinical phenotypes regardless of the inheritance pattern. The classification of CMT has been divided further into subtypes, identified by letters, as defined by the mutated gene (
Table 88.1).
EPIDEMIOLOGY
The prevalence of CMT is about 1 in 2,500 people, with a global distribution and no ethnic predisposition. CMT1A, associated with 17p11.2 duplication in the region containing the peripheral myelin protein 22 gene (PMP22), is the most common form of CMT and accounts for 60% to 70% of demyelinating CMT patients (around 50% of all CMT cases). Mutations in the gap junction beta 1 gene (GJB1) causing CMTX result in approximately 10% to 20% of CMT cases and CMT1B associated with mutations in the myelin protein zero gene (MPZ) accounts for less than 5%. Patients with CMT2 are about 20% of all cases. The prevalence of hereditary neuropathy with liability to pressure palsies (HNPP) is not known, but about 85% of patients with clinical evidence of this syndrome have a chromosome 17p11.2 deletion.
PATHOBIOLOGY
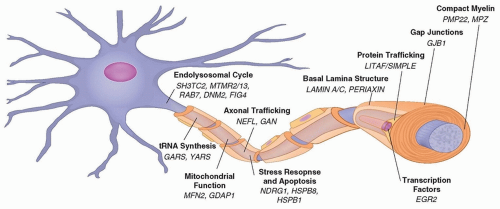
A common feature of most genes mutated in CMT is the role they play in maintaining the structure or function of cellular components of the peripheral nervous system, myelinating Schwann cells, and the axons they ensheath (
Fig. 88.1). Schwann cells and axons interact at multiple points along the peripheral nerve, including the adaxonal (opposing the axon) membrane, paranodal myelin loops, microvilli, and juxtaparanodal basal lamina. These interactions are mutually beneficial, providing trophic support to the axon and myelinating cues to the Schwann cell. An example
of this important interaction is the occurrence of secondary axonal degeneration in all forms of demyelinating CMT. Indeed, although the primary metabolic or structural defect often affects either the myelin or the axon, axonal degeneration represents the final common pathway in both forms of peripheral neuropathies. In demyelinating neuropathies, the secondary axonal degeneration presumably occurs because of inadequate Schwann cell support of the axon. In these neuropathies, secondary axonal degeneration may contribute more to clinical impairment than the primary demyelination. Impaired interactions between axons and mutant Schwann cells seem to be the common process linking all forms of demyelinating CMT. Most cases of CMT1 are caused by mutations in myelin-specific genes including
PMP22 (CMT1A),
MPZ (CMT1B) and
GJB1 (CMT1X), or other genes associated with Schwann cell function, including those that exert transcriptional control of myelination and intracellular trafficking. At a pathologic level, dysmyelination, demyelination, remyelination, and axonal loss are characteristic features of the various demyelinating forms of CMT1. In DSN, myelin may never have formed normally, which is referred to as
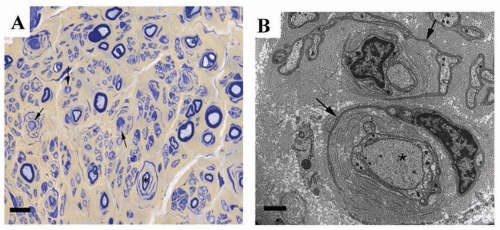
dysmyelination. In CMT1, onion bulbs of concentric Schwann cell lamellae are usually present on nerve biopsies (
Fig. 88.2), with loss of both small- and large-diameter myelinated
fibers and a decrease in the number of myelinated axons. Focal, sausage-like thickenings of the myelin sheath (tomacula) are characteristic of HNPP but may also be found in other forms of CMT1, particularly CMT1B. In CMT1, disability typically correlates better with secondary axonal degeneration than with demyelination itself, again demonstrating the importance of Schwann cell-axonal interactions in demyelinating disease.
Several recent studies have demonstrated a susceptibility of Schwann cells to mutations yielding misfolded proteins, as seen in certain PMP22 and MPZ point mutations. Misfolded proteins may accumulate in the endoplasmic reticulum (ER) of Schwann cells inducing an unfolded protein response (UPR), a series of cellular events that help the ER to cope with the increased metabolic demand caused by retention of the misfolded protein. This, in turn, causes downregulation of the myelination program genes and dedifferentiation of Schwann cells, a toxic gain of function that worsens with the demyelination and is potentially amenable to therapeutic intervention.
Spinal motor neurons and dorsal root ganglion sensory neurons that are affected in CMT have particular challenges in maintaining homeostasis, as their axons extend up to 1 m distal from the cell body. Because the majority of neuronal proteins are synthesized in the cell body, intensive transport of proteins must occur between the soma and the axonal extremity via anterograde transport. Additionally, signals from the periphery containing toxic or prosurvival factors, as well as damaged proteins, return to the cell body via retrograde transport. Indeed, axonal trafficking is emerging as a common theme of various seemingly diverse genes that are associated with CMT type 2. Mutations in genes associated with axonal structure and function result in CMT2. Some examples include mutations in proteins of the neuronal cytoskeleton (neurofilament light chain-NEFL, CMT2E), proteins associated with axonal mitochondrial dynamics (mitofusin 2-MFN2, CMT2A and gangliosideinduced differentiation protein 1-GDAP1, CMT2K), and protein associated with regulation of membrane and intracellular trafficking (Ras-associated protein RAB7, CMT2B, for example). The pathologic hallmark of CMT2 is axonal degeneration with loss of all types of nerve fibers in the absence of onion bulb formation.
CLINICAL MANIFESTATIONS
Despite phenotypic variability, there are characteristic clinical patterns for many types of CMT (
Table 88.2). The “classical” CMT phenotype consists of normal early milestones such as beginning to walk by a year of age. This is followed by gradually progressing weakness and sensory loss during the first two decades of life. The classic phenotype typically features a steppage gait, pes cavus, sensory loss in a stocking or glove distribution, inverted champagne bottle legs, and atrophy in the hands (
Fig. 88.3). Although both motor and sensory nerves are usually affected, the more prominent phenotypic characteristic is related to motor difficulty in most cases. Physical examination shows decreased or absent deep tendon reflexes, often diffusely but always involving the Achilles tendon. Findings are usually symmetric. Pronounced asymmetries in symptoms suggest HNPP if they are episodic. Otherwise, they are more consistent with acquired disorders. Patients with classical CMT almost always have impaired proprioception with balance difficulty. Affected children are usually slow runners and have difficulty with activities that require balance (e.g., skating, walking along a log across a stream). Ankle-foot orthotics (AFOs) are frequently required by the third decade. Fine movements of the hands for activities such as turning a key or using buttons and zippers may be impaired, but the hands are rarely as affected as the feet. Deep and superficial muscles that are innervated by the peroneal nerve, such as the tibialis anterior and peroneus brevis and longus muscles often cause more symptoms than do the plantar flexion muscles innervated by the tibial nerve, such as the gastrocnemius. As a result, tripping and spraining one’s ankle are frequent symptoms. Most patients remain ambulatory throughout life and have a normal life span.
However, CMT, as well as genetically, may be clinically heterogenous, with variability in the age of onset, speed of progression, and electrophysiologic findings. Onset may differ depending on the genetic subtype, including early-onset, infantile forms with delayed milestones (historically designated DSN) and late-onset, adult forms. Symptoms are usually slowly progressive, especially for the classic and late-onset phenotype, but can be severe, particularly in early-onset forms.
ELECTROPHYSIOLOGY
Electrophysiologic studies allow for classification of CMT into demyelinating (CMT1) and axonal (CMT2) forms. The standard cutoff for demyelinating motor nerve conduction velocity (MNCV) is 38 m/s in the upper extremities. Axonal forms (CMT2) exhibit MNCVs greater than 45 m/s but a decrease in compound muscle action potential (CMAP) amplitudes. The dominant intermediate forms (I-CMT) show MNCVs between 25 and 45 m/s. Conduction velocities are performed in the upper limbs because CMAP amplitudes are often unobtainable in the legs, even for demyelinating forms of CMT, due to either conduction failure or secondary axonal degeneration.
The usual electrodiagnostic finding in demyelinated inherited neuropathies is widespread uniform slowing of conduction velocities, as opposed to the multifocal segmental slowing found in demyelinating acquired neuropathies in which temporal dispersion and conduction blocks are frequently seen. Exceptions to this rule are women with CMTX, patients with HNPP, and some CMT1B cases with specific MPZ mutations. In these cases, focal demyelination with temporal dispersion or conduction block can be seen. In other cases of CMT1, the finding of focal slowing should raise the possibility of a superimposed inflammatory neuropathy.