Fig. 15.1
Congenital spinal malformations; disordered embryogenesis during formation of neural tube from neural ectoderm results with myelomeningocele, while different forms of occult dysraphism may occur due to mesodermal disarrangement
The aim of this chapter is to discuss the contemporary treatment protocols of this heterogeneous group with emphasis on changing concepts due to accumulating data on their embryogenesis, natural course, and therapeutic alternatives.
15.1.1 Open Spinal Dysraphism (Myelomeningocele)
Formation of the spinal cord is identified as primary neurulation in classical embryology, wherein ectoderm of the bilaminar embryo undergoes a series of complex differentiation, yet not through a fully understood mechanism, to form the neural tube. This process requires molecular, biochemical, and mechanical interactions between neuroectodermal cells and adjacent ectodermal derivatives [1–4]. Although primary neurulation itself represents neural tissue formation, it is closely dependent on the ongoing mesodermal activity that is responsible for the differentiation of the nonneuronal surrounding tissue. While notochordal induction is essential for initiating primary neurulation, a completed neurulation is required for appropriate differentiation of the surrounding tissue (also see Chap. 14).
15.1.1.1 Pathogenesis
Neural tube formation starts at the 17th day of gestation during which the notochord induces the overlying ectoderm to differentiate into neuroectoderm to form the neural groove in the dorsal midline of the embryo. Neural folds on either side of the groove elevate and meet, the cells fuse dorsally, and a primitive spinal cord is formed as a hollow cylinder. This process is completed by days 27–28 of gestation. Interruption of the neural tube formation dorsally at a particular segment(s) prevents normal neural tissue differentiation at that level. While this maldevelopment of the spinal cord structure causes a more or less complete neurological impairment below the affected level, additional abnormalities appear as a result of altered induction of the neural tube to the surrounding tissues [2, 5]. The consequence is a visible spinal cord segment, placode, representing an unclosed primitive neural tube remnant without meningeal, bony, or cutaneous enclosures dorsally. The term “spina bifida,” although denoting only the missing posterior bony elements over the unclosed neural tissue, is confusingly used to describe the whole anomaly. Likewise, myeloschisis, spina bifida cystica, and myelomeningocele are terms attributed to the different morphological appearance of the same pathology contributing no practical purpose in terms of decision making, surgical technique, or outcome other than confusion. The term “myelomeningocele” is currently preferred to represent almost all variations of open spinal dysraphism that result from a common mechanism of maldevelopment [1, 6, 7].
Disordered embryogenesis of the segmental neural tube formation in myelomeningocele is attributed to either primary failure of neural tube closure or secondary opening after appropriate tube formation. The nonclosure theory is probably for the majority of human myelomeningoceles; however, overdistention may contribute to some experimental neural tube defect models [2, 6]. Regardless of the causative mechanism, unclosed neural tube triggers a cascade of events concerning the nonneural tissue that is designated to cover the spinal cord dorsally. The cutaneous ectoderm remains attached to the open neural tube segment and fails to form the future skin over the lesion. Moreover, the normal mechanism in which the cutaneous ectoderm detaches from the dorsal side of the neural tube to allow paraxial mesoderm to move in between to give rise to bone and soft tissue is also distorted. The final pathological anatomy is an exposed lesion at birth, representing the inner surface of the spinal cord on the dorsal midline covered with membranous tissue as the epidermal remnants or exudate, with a groove continuous with the central canal of the unaffected segments. Immature laminar remnants and paraspinal muscles occupy the lateral border of the widened spinal canal and can be palpated under the border of the skin defect (Fig. 15.2).
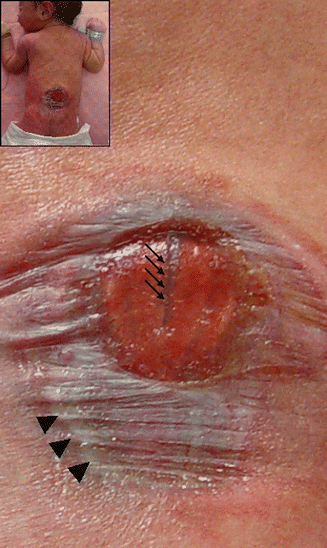
Fig. 15.2
A newborn with myelomeningocele (upper left). Unclosed primitive neural tube remnant, placode, is exposed without any dorsal covering expect for membranes of epidermal remnants or exudate. The groove (arrows) represents the central canal. The healthy skin border (arrow heads) denotes the lateral edges of the widened spinal canal segment
15.1.1.2 Etiology and Epidemiology
Current hypotheses suggest that complex interactions between extrinsic and intrinsic variables are responsible for myelomeningoceles. Clinical and epidemiological data in humans imply maternal illnesses, medications, environmental toxins, and dietary factors such as lack of folic acid that play causative or at least contributing roles in the development of myelomeningocele. On the other hand, increased incidence demonstrated in certain families situates neural tube defects into complex genetic disorders in which genes and the environment interact through an unknown relationship [2–4].
The incidence of myelomeningocele is generally accepted as 1/1000 live births regardless the ethnic and geographic variability. Although several studies demonstrate a variation in different parts of the world depending on the geographical region, seasons at conception, gender of the affected infants, ethnicity, and socioeconomic status of the parents, maternal age and parity and population-based surveillance studies fail to confirm a definitive correlation with the incidence [8]. A decrease in frequency of myelomeningocele has been reported recently in some areas, while the incidence has been stable elsewhere [9]. Although this decrease has been attributed to increased prenatal diagnosis, selective terminations, genetic counseling, and mostly folic acid supplementation during pregnancy, there are no hard data to indicate that the decrease is due to a single factor [10, 11].
15.1.1.3 Presentation
Myelomeningocele represents one of the most devastating congenital malformations that are compatible with life. This is due to the fact that neurological impairment is inevitable for a patient with myelomeningocele and the severity is proportional to the affected level of the spinal cord. With up to 80 % of the disclosure taking place at the thoracolumbar spine, paraplegia is the result. The level of neurological deficit descends as the lesion level moves caudally; at best, sacral localization avoids a major motor disturbance but does result in a neurogenic bladder [7, 12, 13].
The neurological deficit in myelomeningocele is thought to be due not only to incomplete differentiation of the neural tube but also to exposure of the uncovered neural tissue to amniotic fluid. Furthermore, associated anomalies extending to 63 % as reported in fetal autopsy series contribute to the disability of the myelomeningocele cases [3]. Besides morphological abnormalities of the adjacent vertebral elements, almost all patients with myelomeningocele have associated Chiari II hindbrain malformation. The simplest representation of Chiari II malformation is herniation of the cerebellar tonsils and vermis to the cervical spinal canal through a tight foramen magnum. Additionally, medullary kinking, low-lying tentorium, tectal beaking, brain stem nuclei changes, polymicrogyria, and gray matter heterotopias may be associated with the Chiari malformation. The main contribution of the tonsil herniation to the clinical picture of myelomeningocele is hydrocephalus, which is present in almost 90 % of patients either during delivery or becoming apparent after surgical treatment [1, 12]. Hydrocephalus are one of the major coexisting factors that are responsible for morbidity and overall unfavorable outcome in myelomeningocele cases. Craniolacunae, a mesodermal self-limiting skull abnormality, is also a frequent finding in the newborn.
From the practical viewpoint, a patient with myelomeningocele is born with signs of functional cord transection at the lesion level and neurogenic bladder, and has a very good chance of hydrocephalus. The open lesion carries a substantial risk of getting infected; cerebrospinal fluid exposure to the external environment through the incomplete dural barrier can initiate meningitis and ventriculitis. Meningitis not only complicates a probable treatment for hydrocephalus but also adds the potential risk of seizures and further neurological impairment in terms of intellectual outcome.
15.1.1.4 Surgical Treatment
The aim of surgical treatment for myelomeningocele is to stabilize the clinical and neurological status of the newborn and prevent the potential risks of deterioration. This is best achieved by reconstructing the open neural tube and its coverings as soon as possible after birth. The initial management aims to stabilize the infant, avoiding contamination of the lesion and excluding the associated malformations. First, the important concern at this point is the decision to treat. This has medical, ethical, and legal ramifications to be discussed among the parents and the physicians. In cases with a prenatal diagnosis, the parents have already acknowledged the consequences of a congenital anomaly and treatment options. Otherwise, the anticipated problems in myelomeningocele with the limited role of surgical repair in the final outcome may result in the refusal of surgical treatment. From the physicians’ side, severe forms with multisegment large lesions complicated with associated vertebral anomalies and hydrocephalus may cause hesitation to treat. Unless there is a life-threatening coexisting malformation, the current ethicolegal opinion is to provide surgical treatment to all cases [6, 7, 12]. Once treatment has been decided, the second concern is the timing of the surgical procedure. Risks of immediate repair in a newborn should be weighed against the risk of contamination at delayed closure. Surgical repair within 48–72 h after birth is universally accepted and does not necessarily carry increased risk of contamination compared to very early treatment (within the first 24 h). Furthermore, the time interval provides sufficient postnatal evaluation and stabilization of the infant. The simplest description of the surgical technique is to mimic the normal embryological pattern of development. This is to isolate the nonfused segment, establish the original tube shape for the placode, and recreate and close the dural envelope followed by approximation and closure of the skin over the lesion (Fig. 15.3a–d). Even very large defects can be closed by relaxation incisions along the axis, avoiding complex muscle and skin flaps once advocated but proved to have major consequences. Almost 10 % of cases exhibit hydrocephalus at birth, necessitating simultaneously shunting with repair of the spinal defect. Operative mortality is nearly zero, but major morbidity includes progressive hydrocephalus, wound infection, breakdown, and leakage of cerebrospinal fluid.
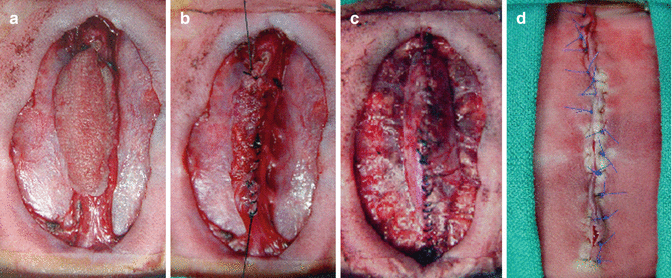
Fig. 15.3
Basic steps of myelomeningocele closure. (a) Removing membranes and debris and exposure of the placode, (b) reapproximation of the flat placode to a tubular form with fine sutures for an easy closure, (c) detaching the dural layer from paravertebral fascia and watertight closure over the exposed neural tube, and (d) primary closure of the skin defect
Following myelomeningocele repair, the treatment of the other conditions may range from simple observation to extensive surgical procedures. The vast majority of patients will require shunts for hydrocephalus before being discharged; future treatment might be required for associated kyphosis, Chiari malformation, foot deformities, and secondary tethering of the spinal cord for CM-II, syringomyelia, and/or tethered cord syndrome. While myelomeningocele may be regarded a static and nonprogressive defect, clinical worsening is caused by associated problems. Owing to the fact that myelomeningocele is located at thoracolumbar segments in almost 80 % of the cases, the child faces lifetime complications of paraplegia and neurogenic bladder. Therefore, the initial closure is just the beginning, and outcome and long-term results greatly depend on the management of the associated conditions [7, 12]. At least 75 % of children born with an open spina bifida can be expected to reach their early adult years. Survivors have a high incidence of problems related to pressure sores, obesity, severe renal disease, hypertension, depression, and visual impairment. Mortality is mostly related to shunt dysfunction and infection, urinary complications of neurogenic bladder, or respiratory tract infection [13].
Fetal Myelomeningocele Repair
Fetal surgery is routinely performed for various conditions, and myelomeningocele is also a good candidate for in-utero repair since it is associated with considerable morbidity after postnatal care, it is compatible with life, and can be detected before the 20th week of gestation. Furthermore, there is enough experimental evidence that some function of the placode is preserved initially but can deteriorate during gestation or at birth. Fetal closure of the open neural tube can prevent secondary damage and preserve neurological function, while existing fetal potential for wound healing and axonal regeneration might reverse preexisting injuries to a certain degree. Since 1994, more than 330 cases of intrauterine repair have been performed by standard multilayer reconstruction through a hysterotomy between 19 and 25 weeks gestation in certain centers worldwide. Preliminary findings suggested that intrauterine myelomeningocele repair may lessen the degree of Chiari malformation and reduce the incidence of shunt-dependent hydrocephalus [14]. Therefore, a multicenter randomized controlled trial was initiated in the United States to compare intrauterine with conventional postnatal care in order to establish the procedure-related benefits and risks. The Management of Myelomeningocele Study (MOMS) was terminated early in 2010 due to the efficacy of prenatal surgery. The researchers found that intrauterine repair was associated with a 52 % risk reduction with regards to shunt placement and at 30 months the patients in the prenatal group had better motor function and were more likely to walk without an orthotic device [15]. These findings are especially important since the anatomical lesion in the prenatal group was significantly higher. However, the benefits of prenatal repair of myelomeningocele must be weighed against a higher rate of prematurity and maternal morbidity.
Type 1 Chiari malformation is the most common neural axis abnormality in infantile and juvenile idiopathic scoliosis [16]. Furthermore, between 50 and 75 % of these patients will have concomitant syringomyelia, and anywhere from 40 to 80 % will have scoliosis. The incidence of syringomyelia-associated scoliosis falls to less than 20 % for patients over the age of 20 [17]. As compared to adolescent idiopathic scoliosis, this patient population tends to have more atypical curve patterns with a higher curve apex, but a right thoracic remains the predominant curve [17, 18]. The severity of the spinal deformity, however, has not been shown to be correlated with the length of the syringomyelia cavity.
It remains prudent to first perform a Chiari decompression, with or without duraplasty, since this has been associated with a decrease in the size of the syrinx and possible improvement or stabilization of the spinal curvature. Suboccipital decompression is preferred over shunting, as the benefits on curve progression have not been observed when shunting of the syrinx is performed alone [19]. Likewise, the possible benefits of neurosurgical intervention do not extend to patients with congenital scoliosis and syringomyelia.
In several studies of children under the age of 10, over 90 % experienced an improvement or stabilization of their spinal curve following Chiari decompression [16, 17]. Although others have reported more modest results following decompression, averaging about 50 %, there appears to be a consensus in the literature that patients who progress tend to be older and have larger, double curves [17, 19, 20]. It is important to note that more extensive decompression of the cervical spine, laminectomies below C1, and extensive muscle stripping have been reported to worsen the spinal deformity, especially in the sagittal plane [18]. Finally, the effects of decompression may be temporary; hence, a follow-up period of at least 5 years is advised [19].
15.1.2 Closed (Occult) Spinal Dysraphism
Occult spinal dysraphisms represent a wide spectrum of malformations within congenital spinal disease, the only common feature being a mesodermal developmental error covered with normal skin [21]. Unlike the neuroectoderm that is designated to initiate the spinal cord, embryonic mesoderm gives rise to a variety of structures. Any disarrangement during the differentiation of this pluripotent layer triggers diverse forms of disease with regard to anatomy, clinical presentation, and treatment options. While the neurological impairment in open dysraphism is straightforward related to the incomplete differentiation of the neural tissue, the mechanism of neurological consequences in closed forms are far more complex and controversial. This complexity, in turn, generates an ongoing controversy in establishing universal algorithms for treatment.
15.1.2.1 Pathogenesis
Neurulation is responsible for the formation of the spinal cord until the future second sacral segment and the most distal segment of the spinal cord develop by a process called secondary neurulation from the neural ectoderm cell mass caudal to the neural tube, the caudal eminence. Caudal eminence is formed from pluripotent cells derived from the regressing primitive streak. The mesenchymal neural cord then becomes an epithelial cord, acquires a lumen by canalization and regression process, attaches to the primary neural tube, and forms the remaining sacral and coccygeal segments of the spinal cord including the filum terminale [1, 3, 22]. Developmental errors during secondary neurulation, besides several anomalies, lead to the formation of a fatty and short/thick filum, a classical representative of occult spinal dysraphism. Other major occult forms of dysraphism include split cord malformations (SCMs, diastematomyelia), lipomyelomeningoceles, and dermal sinuses which represent disordered mesodermal differentiation belonging to different stages during primary neurulation, before secondary neurulation begins [23]. The setoff time for the mesodermal maldevelopment and the stage of neurulation at that instance are critical for the neurological consequences. The more the primary neurulation is disrupted, the chances are higher that the child is born with a neurological compromise. This is one of the main reasons why, in different forms of occult dysraphism, resultant neurological status ranges from normal to severe impairment, sometimes compatible with myelomeningocele. Segmental, asymmetrical involvement of neurulation results with lower extremity changes, including leg or buttock asymmetry, hip and knee problems, and foot deformities that typically worsen due to muscle imbalance, weight bearing, and gravity as the child grows [6].
Related posts:
 Other Neuromuscular Diseases
Other Neuromuscular Diseases
 Clinical Examination and Associated Comorbidities of Early Onset Scoliosis
Clinical Examination and Associated Comorbidities of Early Onset Scoliosis
 Revision Spine Surgery in the Growing Child
Revision Spine Surgery in the Growing Child
 Convex Growth Arrest for Congenital Scoliosis
Convex Growth Arrest for Congenital Scoliosis
 Growth-Guided Instrumentation: Shilla Procedure
Growth-Guided Instrumentation: Shilla Procedure
 Plate-Rod System in the Management for Progressive Scoliosis in Growing Children
Plate-Rod System in the Management for Progressive Scoliosis in Growing Children
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


