Metabolic myopathies comprise a clinically and etiologically diverse group of disorders due to defects in cellular energy metabolism, including the breakdown of carbohydrates and fatty acids to generate adenosine triphosphate (ATP), predominantly through mitochondrial respiratory chain oxidative phosphorylation. Accordingly, metabolic myopathies can be etiologically classified into three broad categories: glycogen storage diseases (GSDs), fatty acid oxidation (FAO) disorders, and mitochondrial diseases. These metabolic myopathies present with a clinical spectrum ranging from severe infantile-onset multisystemic diseases to mild adultonset isolated myopathies. In this chapter, we focus on metabolic myopathies that affect adults.
EPIDEMIOLOGY
Metabolic myopathies are rare diseases. The most common of these disorders is Pompe (acid maltase deficiency) disease with variable ethnic and geographic incidence from 1:14,000 births annually in African-Americans to 1:100,000 in people of European descent. Prevalence of late-onset Pompe disease has been estimated to be 1:60,000. Other frequent metabolic myopathies include McArdle disease (myophosphorylase deficiency) and carnitine palmitoyltransferase II (CPT II) deficiency. The prevalence of McArdle disease has been estimated to be 1:100,000 in the Dallas-Fort Worth, Texas region and a minimum of 1:170,000 in Spain. The prevalence of CPT II deficiency is unknown; however, over 300 patients have been reported. Although individual mitochondrial diseases are rare, collectively, the prevalence of mitochondrial disorders in adults is approximately 1:5,000.
PATHOBIOLOGY
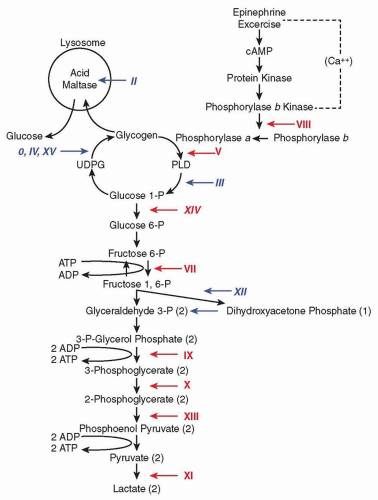
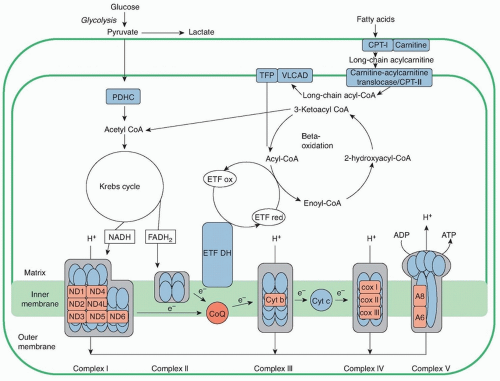
Energy in the form of ATP is required to drive numerous cellular functions including muscle contraction. The main fuels used to generate ATP are glycogen, glucose, and free fatty acids (FFAs). Glycogen is metabolized in the cytoplasm to pyruvate, which enters mitochondria (Fig. 95.1). Short-chain and medium-chain fatty acids cross freely into the mitochondria, whereas long-chain fatty acids require binding to carnitine for transport across the mitochondrial membrane, a process mediated by acyl-carnitine translocase and CPTs I and II (Fig. 95.2). Once in the mitochondria, all these substrates are turned into acetyl-coenzyme A (CoA), which feeds into the Krebs cycle. In this critical cycle, reducing equivalents (electrons) combined with protons are bound to intermediate molecules, nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide, reduced (FADH2), which deliver the electrons to the mitochondrial respiratory chain to produce ATP and water (H2O).
Defects in any of these pathways—glycogen catabolism (glycogenolysis and glycolysis), fatty acid oxidation, Krebs cycle, and mitochondrial respiratory chain and oxidative phosphorylation—cause human disorders that often predominantly affect muscle due to its high energy requirements, particularly during exercise. Because most of the enzyme defects are partial, many of these diseases manifest in adulthood with muscle symptoms either in isolation or with other clinical features.
Abundant glucose is stored in liver and skeletal muscle in the form of a polysaccharide called glycogen. The glycogenoses include disorders characterized by genetic mutations in glycogen synthesis (glyconeogenesis), degradation (glycogenolysis), or glucose degradation (glycolysis). To date, 15 glycogenoses have been identified (see Fig. 95.1); most are autosomal recessive except for phosphoglycerate kinase (PGK) and phosphorylase b kinase deficiency, which are X-linked.
Fatty acids are the primary energy source for muscle at rest and during periods of prolonged low-intensity exercise. Fatty acids are catabolized by the β-oxidation enzymes, which cleave two-carbon fragments with each cycle. Thus, lipidoses arise due to failure of fatty acid transport into mitochondria secondary to carnitine or CPT I or II deficiencies or to defects of intramitochondrial β-oxidation. These disorders are inherited as autosomal recessive traits. The more severe variants present in infancy or childhood with primary involvement of liver or brain, whereas the milder adult forms are predominantly myopathic and include the myopathic variant of CPT II deficiency, trifunctional protein deficiency (TFP), and very long chain acyl-CoA dehydrogenase deficiency (VLCAD).
In addition to catabolizing lipids, mitochondria perform additional essential functions including Krebs cycle and amino acid metabolism as well as energy production through the respiratory chain and oxidative phosphorylation. The respiratory chain is composed of four multisubunit enzymatic complexes (I, II, III, and IV), which generate a proton gradient across the inner mitochondrial membrane (IMM) that, in turn, drives ATP synthesis by complex V. In addition, CoQ10 and cytochrome c are critical components of the mitochondrial respiratory chain, serving as “electron shuttles” between the complexes. Originally restricted to primary defects of the respiratory chain and oxidative phosphorylation, mitochondrial diseases have expanded to encompass defects of mitochondrial transcription, translation, protein importation, lipid membranes, and organellar dynamics (fusion, fission, and movement) (see Chapter 139).
CLINICAL FEATURES
From a clinical point of view, metabolic myopathies can be categorized into two different groups: (1) those that show symptoms and signs related to exercise (exercise intolerance, cramps, myalgias, myoglobinuria) with normal interictal examination and (2) those with fixed symptoms such as muscle weakness often associated with systemic involvement (such as encephalopathies or endocrinopathies). When evaluating a patient with exercise-related symptoms, clinicians should ask two questions: (1) What type of exercise provokes symptoms? (2) Are there associated triggering factors? If short bursts of high-intensity exercise cause muscle cramps or myoglobinuria, the patient may have a defect of glycogen metabolism. Examples of this type of activity include weight lifting or sprinting. In young patients who play baseball or softball, the “home run” sign (Haller sign), inability to sprint around the bases due to exercise-induced muscle spasms, is a typical complaint in patients with glycogenoses, such as McArdle disease. In contrast, if the patient complains that prolonged exercise (such as hiking or playing soccer) triggers myalgias, fatigue, and myoglobinuria without acute contractures, the patient is likely to have a defect of fatty acid oxidation. The symptoms often occur when the patient is fasting or under stress. A prototypical example is a young adult with CPT II deficiency who enlists in military service and has difficulty completing long marches due to fatigue and myalgias followed by myoglobinuria.
FIGURE 95.1 Scheme of glycogen metabolism and glycolysis. Roman numerals denote muscle glycogenoses due to defects in the following enzymes: 0, glycogen synthase; II, acid maltase; III, debrancher; IV, brancher; V, myophosphorylase; VII, phosphofructokinase; VIII, phosphorylase b kinase; IX, phosphoglycerate kinase; X, phosphoglycerate mutase; XI, lactate dehydrogenase; XII, aldolase; XIII, β-enolase; XIV, phosphoglucomutase 1; and XV, glycogenin-1. Red numerals designate glycogenoses associated with exercise intolerance, cramps, and myoglobinuria. Blue italic numerals correspond to glycogenoses causing weakness.
In the past, the term myoglobinuria was reserved for grossly pigmented urine, but modern techniques can detect amounts of this protein so minute that discoloration may not be evident. (Determination of serum myoglobin content by radioimmunoassay has similar diagnostic significance as measurement of serum creatine kinase [CK] activity.) The clinically important syndromes, however, are associated with gross pigmenturia. Numerous conditions cause myoglobinuria (Table 95.1). Sometimes, the disorder can be recognized without direct demonstration of myoglobin in the urine: For instance, acute renal failure in a patient with levels of serum CK activity over 1,000 units would suggest myoglobinuric renal failure. Inexplicably, the cumbersome neologism rhabdomyolysis was favored for a few years, but a 2015 Medline search found that “myoglobinuria” is holding fast. Myoglobin is the visible pigment in the urine, and it is a toxin that injures the kidney; the syndrome originates with muscle necrosis, which does not need a new name.
Some patients with glycolytic, lipid, or mitochondrial disorders can develop isolated progressive myopathy and persistent weakness. More typically, patients with mitochondrial diseases can show a wide range of extramuscular manifestations. In this section, general clinical aspects of these metabolic myopathies with emphasis on the more common forms are described.
DISORDERS OF GLYCOGEN METABOLISM (GLYCOGENOSES)
Clinical presentations of muscle glycogenoses are protean, ranging from profound multisystem disease in infancy to exercise intolerance or isolated progressive muscle weakness in adults. Here, we focus on the adult patients. Myophosphorylase deficiency (McArdle disease, glycogenosis type V) is a prototypical glycogenolytic disorder with episodic muscle dysfunction and myoglobinuria. It is the most common disorder of skeletal muscle carbohydrate metabolism and one of most frequent genetic myopathies. Patients with McArdle disease typically exhibit intolerance to static or isometric muscle contractions and also to dynamic exercise that can trigger episodes of reversible “muscle crises.” Acute crises manifest mainly in the form of premature fatigue and contractures, frequently accompanied by muscle breakdown (rhabdomyolysis) with elevated serum CK and sometimes by myoglobinuria. In addition to McArdle disease, five other forms of glycogenoses manifest exercise-induced myoglobinuria: type VII (phosphofructokinase [PFK] deficiency, Tarui disease), type VIII (phosphorylase b kinase), type IX (PGK deficiency), type X (phosphoglycerate mutase [PGAM] deficiency), type XI (lactate dehydrogenase deficiency), and type XIV (phosphoglucomutase 1 deficiency).
FIGURE 95.2 Schematic representation of mitochondrial metabolism. CPT, carnitine palmitoyltransferase; TFP, trifunctional enzyme; VLCAD, very long chain acyl-CoA dehydrogenase; PDHC, pyruvate dehydrogenase complex; CoA, coenzyme A; ETF ox and red, electron transfer flavoprotein oxidized and reduced; NADH, nicotinamide adenine dinucleotide; FADH2, flavin adenine dinucleotide, reduced; ETF DH, electron transfer flavoprotein dehydrogenase; CoQ, coenzyme Q; Cyt c, cytochrome c.
Another important sign considered pathognomonic is the “second wind,” which is a marked improvement in exercise tolerance about 10 minutes into aerobic exercise involving large muscle masses (jogging or cycling). The second wind, as manifested by a marked decrease in early exertional tachycardia (e.g., a decrease from ˜140 to 150 beats per minute to ˜120 beats per minute) starting after around 7 minutes of exercise, does not occur in patients with other disorders that are also associated with exercise intolerance, such as PFK deficiency and other glycogenoses, mitochondrial myopathies, or disorders of lipid metabolism. This phenomenon is due to increased uptake of glucose and use of fatty acid.
Hemolytic anemia (elevated indirect bilirubin and reticulocytes) is seen in glycogenosis due to defects in genes partially expressed in erythrocytes such as PFK, PGK, and aldolase A. Cognitive deficits are often associated with the adult polyglucosan body disease (APBD) form of branching enzyme deficiency (type IV) and PGK deficiency.
Acid maltase or acid α-glucosidase (GAA) is an enzyme responsible for the catabolism of glycogen within lysosomes. Infantileonset acid maltase deficiency (type II, Pompe disease) presents as a myopathy and cardiomyopathy, which, if untreated, is typically fatal in the first year of life. In contrast, in the late-onset form of Pompe disease beginning in childhood through adulthood, patients have slowly progressive fixed proximal muscle weakness and early respiratory insufficiency. Although myopathy is the predominant manifestation, patients with Pompe disease also develop basilar artery and aortic aneurysms, bladder or bowel incontinence, and dysphagia. An autopsy study of a late-onset Pompe patient revealed ultrastructural abnormalities in the smooth muscle of blood vessels, gastrointestinal tract, and bladder, thereby accounting for the extraskeletal muscle manifestations. Hearing loss has also been reported in this disease. It is important to diagnose Pompe disease, as enzyme replacement therapy with recombinant human GAA (rhGAA) dramatically improves the cardiomyopathy in the infantile form and less effectively improves the myopathy in both infantile and late-onset forms.