MS is a chronic inflammatory demyelinating disease of the central nervous system (CNS) of unknown cause. The course is extremely variable, but most patients initially experience relapses with complete or near-complete recovery interspersed with periods of clinical remission. Although a minority of patients has only minimal symptoms, many become disabled in time as a result of incomplete recovery from relapses or conversion to a progressive form of the disease.
PATHOBIOLOGY
The etiology of MS is unknown. It probably results from complex interactions between environmental factors and susceptibility genes, which lead to an aberrant immune response and damage to the myelin sheath, oligodendrocytes, axons, and neurons.
Studies in a mouse model of MS, experimental autoimmune encephalomyelitis (EAE), histopathologic studies of MS lesions, and immunologic markers in serum and cerebrospinal fluid (CSF) of MS patients suggest that MS is an immune-mediated disease. A virus, bacterium, or other environmental toxin might induce an immune response in genetically susceptible persons. Antigen-presenting cells (APCs) provide relevant antigens to CD4+ T-helper cells in the periphery, which lead to their activation and the subsequent generation of autoreactive proinflammatory T-helper (Th) 1 and 17 subsets. B cells and monocytes are also activated. These autoreactive T cells interact with adhesion molecules on the endothelial surface of CNS venules and, with antibodies and monocytes, cross the disrupted blood-brain barrier with the aid of proteases (e.g., matrix metalloproteinases) and chemokines. Within the CNS, target antigens are recognized (putative antigens include myelin basic protein, myelin-associated glycoprotein, myelin-oligodendrocyte glycoprotein, proteolipid protein [PLP], αB-crystallin, phosphodiesterases, and S-100 protein), T cells are reactivated, and the immune response is amplified. Proinflammatory T-helper cells proliferate and B cells continue their maturation to antibody-secreting plasma cells, whereas monocytes become activated macrophages. Together, these immune cells produce inflammatory cytokines (e.g., interleukin [IL]-12, IL-23, interferon γ, tumor necrosis factor [TNF]-α), proteases, free radicals, antibodies, nitric oxide, glutamate, and other stressors that collectively lead to damage of myelin and oligodendrocytes. In the appropriate cytokine milieu, CD4+ Th2 cells proliferate and secrete anti-inflammatory cytokines (e.g., IL-4, Il-5, IL-13, and transforming growth factor-β) that suppress the immune response. Depending on the location and extent of damage, demyelination may impair or block nerve conduction and result in neurologic symptoms. With a loss of trophic support from oligodendrocytes, axons may degenerate to cause irreversible neurologic deficits. Spontaneous improvement of symptoms is attributed to resolution of inflammation, adaptive mechanisms (e.g., reorganization of sodium channels), or remyelination.
It had long been thought that Th1 and Th2 subsets arose from the terminal differentiation of the CD4+ T cells. However, a third pathway has been identified, induced by IL-1, IL-6, and transforming growth factor-β, and then expanded and maintained by IL-23, which is secreted by APCs. This third subset, a proinflammatory T-helper cell, is known as Th17 because it produces IL-17. Th17 cells secrete a number of cytokines, including TNF-α and granulocyte macrophage colony stimulating factor (GM-CSF), which are critical for the development of EAE. MS patients have monocyte-derived dendritic cells that secrete higher levels of IL-23 than healthy controls. Higher levels of IL-17 mRNA-bearing mononuclear cells are found in the serum of MS patients having relapses than in patients in remission.
Although MS is typically considered a T-cell-mediated disease, a growing body of evidence supports a pathogenic role of B cells, including the frequent observation of intrathecal production of immunoglobulin in MS patients, identification of antibodies that react to specific myelin antigens within MS lesions, a pathologic pattern of MS characterized by antibody-associated demyelination (see the following text), and the discovery of B-cell follicles in the meninges of patients with secondary progressive MS. Pathologic studies have shown that B-cell clones are shared between the meninges and parenchyma of individual MS patients. Furthermore, B cells
are efficient APCs and B-cell depletion is a promising therapeutic approach in MS.
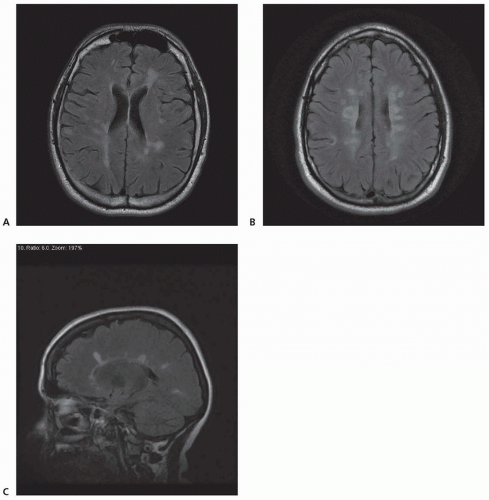
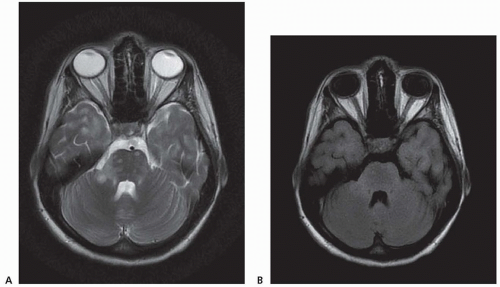
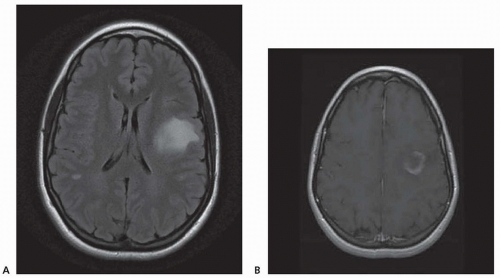
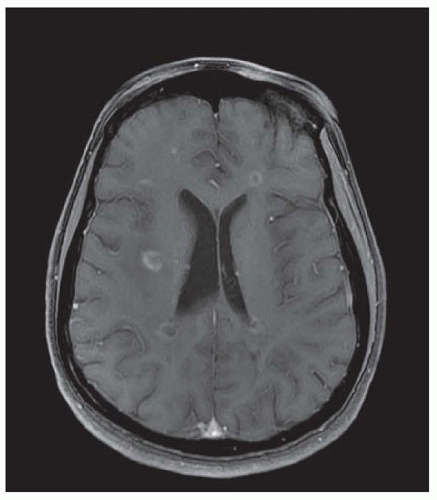
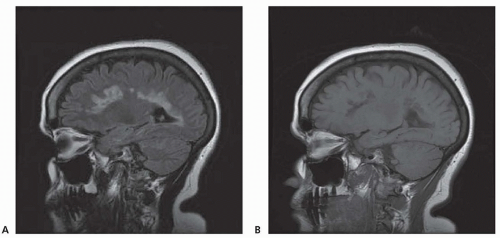
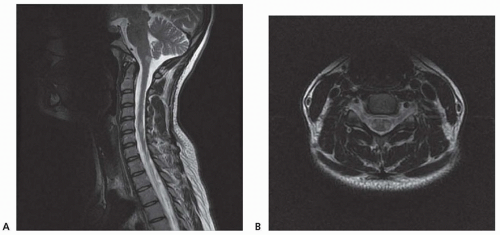
Chronic demyelinating plaques appear translucent, sharply demarcated, and are most frequently found in the periventricular white matter, brain stem, cerebellum, and spinal cord. Lesions are characterized by extensive demyelination, gliosis, variable axonal loss, and a minimal inflammatory infiltrate consisting of T lymphocytes and macrophages. Demyelination accompanied by a perivascular infiltrate consisting predominantly of T cells, lipidladen macrophages, and prominent reactive astrocytes are typical features of actively demyelinating lesions. Although demyelination with relative preservation of axons is often considered the pathologic hallmark of MS, axonal transection is common, especially in areas of active inflammation and demyelination. In autopsies of 52 MS cases, prominent cortical demyelination was seen with a mild but diffuse inflammatory infiltrate with microglial activation in normal-appearing white matter of patients with secondary or primary progressive MS. Cortical demyelination was rare in patients with relapsing-remitting MS.
Lucchinetti and colleagues identified four distinct pathologic patterns in an immunohistopathologic study of actively demyelinating MS lesions in 83 cases (51 biopsies and 32 autopsies). All four patterns contained an inflammatory infiltrate consisting of T lymphocytes and macrophages. The most common type, pattern II, was characterized by the deposition of immunoglobulin and complement. Pattern I was characterized by macrophageassociated demyelination. In patterns III and IV, demyelination was due to an oligodendrogliopathy. Pattern III was differentiated from pattern IV by a preferential loss of myelin-associated glycoprotein. Multiple active lesions were discovered in 27 autopsy cases. The same lesion pattern was observed within each patient, but there was marked heterogeneity between patients, suggesting that MS might have multiple pathogenic mechanisms. However, the results of this study were challenged by another autopsy series of 12 relapsing-remitting MS patients who died in the setting of an acute relapse. Most cases, including one who died 17 hours after an exacerbation, included lesions characterized by extensive oligodendrocyte apoptosis with intact myelin sheaths and slight or no inflammatory infiltrate. More than one of the aforementioned patterns were observed in some patients. The investigators concluded that pattern III lesions represent an early stage of lesion formation that precedes inflammation and demyelination. However, biopsy and some autopsy studies are susceptible to inherent selection bias and the findings may not be representative of typical MS.
Genetic Risk Factors
The strongest known genetic factor influencing MS susceptibility is the human leukocyte antigen (HLA)-DRB1*1501 haplotype. However, it is not essential for the development of MS, increases the risk two- to fourfold, and is present in 20% to 30% of normal individuals. Linkage and association analysis of 931 family trios (individuals with MS and their parents) screened 300,000 single-nucleotide polymorphisms and identified two genes outside of the HLA region, interleukin-2 receptor alpha gene (IL2RA) and interleukin-7 receptor alpha gene (IL7RA), which also increase the risk of MS. IL2RA encodes the alpha chain of the IL-2 receptor, which is essential for regulation of T-cell responses and has been implicated in the pathogenesis of other autoimmune diseases, including Graves disease and type 1 diabetes mellitus. IL7RA encodes the alpha chain of the IL-7 receptor. IL-7 functions in the homeostasis of memory T cells and may play a role in the generation of autoreactive T cells in MS patients. The effect of these allelic variants on overall MS risk is small but statistically significant. Additional genome-wide association studies have identified 110 MS risk variants in 103 loci, which are not within the major histocompatibility complex.
Additional evidence for a genetic predisposition includes increased risk in some ethnic groups (e.g., Caucasians of northern European ancestry) and decreased risk in others (e.g., Native Americans), varying prevalence rates among different racial groups in the same geographic location, a 20% to 40% increased risk of MS in first-degree relatives, whereas adopted children of MS patients have a risk similar to the general population, and 25% to 30% concordance in monozygotic twins compared to 5% in dizygotic twins. However, 70% of identical twins are discordant for MS, so environmental factors and other unknown influences must contribute to susceptibility.
Environmental Influences
In general, there is a latitudinal gradient with an increased prevalence of MS further from the equator in both hemispheres. Large differences in the frequency of MS are observed in some homogenous populations living at different latitudes. Several regions with similar latitude have vastly different MS prevalence rates, which in some instances can be accounted for by differences in ethnic susceptibility (e.g., Great Britain and Japan lie at the same latitude, but the prevalence in Britain is approximately 60 times more than in Japan).
Further evidence of an environmental effect comes from migration studies and apparent epidemics and disease clusters. In general, immigrants who move from one area to another before the age of 15 years acquire the MS prevalence rate of the new region. The altered risk develops gradually and may be imperceptible in the immigrants themselves but is obtained by their children. Notable exceptions include Israeli-born children of European and American immigrants who have a frequency of MS similar to that of their parents. Therefore, genetics may sometimes be more important than the environment. Migrating after age 15 years does not affect the risk of MS.
Apparent MS epidemics occurred in Iceland and the Faroe Islands after World War II, suggesting an infectious or other environmental cause. The arrival of British troops to the Faroe Islands in 1940 also brought the first neurologist to the islands, so that increased awareness of the disease and better case ascertainment may have led to the apparent “epidemic.” However, the onset of the disease occurred after 1942 in all of the identified cases except for a few who had spent time away from the islands.
More than the expected number of MS cases has been reported in several areas, including Key West, Florida; Orange County, California; Los Alamos County, New Mexico; Hordaland, Norway; and Colchester County, Nova Scotia, raising the possibility of a shared environmental exposure causing the disease. Mercury toxicity, zinc, viruses, and other toxins have been suggested but no credible evidence has explained these clusters, which may have occurred by chance.
Multiple environmental factors may influence the risk of MS, including vitamin D, which has emerged as one compelling possibility. The active form of vitamin D, 1,25-dihydroxyvitamin D
3, has immunomodulatory properties and can prevent or ameliorate experimental allergic encephalomyelitis (EAE), a mouse model of MS. Numerous studies have found an inverse correlation between sunlight exposure (the most common source of vitamin D) and dietary intake of vitamin D and the risk of MS. The relationship to sunlight exposure may help explain the latitudinal variation of MS prevalence. Furthermore, a study of U.S. military personnel found that higher serum levels of 25-hydroxyvitamin D
3 (25[OH] D
3) were associated with a lower risk of MS. Similar findings in the
Netherlands were reported, but only in women, as well as a negative correlation between 25(OH)D
3 levels and disability. However, it is not known whether vitamin D supplementation modifies the disease in people already diagnosed with MS.
High-salt diet has been shown to increase severity of EAE and induce pathogenic TH17 cells in humans and mice in vitro. This has been hypothesized to explain in part the increasing incidence of MS and other autoimmune diseases in countries with “Western diet.”
Among the possible infectious causes, the case for EBV is of interest. The frequency of MS is low in people who are seronegative for EBV, but the risk is increased in those who have had infectious mononucleosis. The evidence for other microbial agents is less compelling, but it is possible that a number of viruses or bacteria might act as a nonspecific trigger of MS in genetically susceptible individuals.
Physical trauma or psychological stress might precede the onset of either MS or MS exacerbations. Although temporal associations are common, a clear causal relationship is lacking. The Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology concluded that there is no significant association between trauma and MS onset or exacerbation. The committee also concluded that, although possible, there is no clear relationship between antecedent psychological stress and the onset of MS or exacerbations. However, a study in Israel concluded that stress associated with the 2006 war between Israel and Lebanon increased exacerbations in civilians with MS.
CLINICAL MANIFESTATIONS
A clinically isolated syndrome (e.g., optic neuritis, transverse myelitis, or a brain stem or cerebellar syndrome) heralds the onset of the disease in approximately 85% to 90% of all cases. A relapsing-remitting course ensues and is often followed by a progressive phase of the disease. Symptoms, which depend on lesion location and extent of tissue destruction, range from mild and intermittent to severe and persistent or progressive (
Table 69.1). The disease begins insidiously and gradually worsens in the remaining 10% to 15% of patients, termed
primary progressive multiple sclerosis.
Acute optic neuritis is one of the most common manifestations. Patients typically note unilateral visual loss that evolves over a few days and is preceded or accompanied by orbital pain that occurs with or is exacerbated by eye movement. Progressive visual failure over many years is uncommon. On examination, visual loss varies from mild to severe. Visual acuity of 20/200 or worse is found in about 33% of patients, but complete loss of vision is rare. Dyschromatopsia and a central scotoma or other visual field defects are common. A relative afferent pupillary defect (RAPD), which may be the only clinical evidence of optic neuritis, is invariably present unless there is a prior history of or current optic neuritis in the opposite eye. The absence of a RAPD in a person with acute visual loss raises the possibility of uveitis, which occurs with increased frequency in MS. Funduscopy in optic neuritis is often normal, but swelling of the optic disc is observed in 35% of patients. Retinal hemorrhages and exudates are uncommon.
Spontaneous recovery of vision usually occurs within the first month, even when visual loss is severe. Lack of improvement suggests an alternative diagnosis because 98% of patients with visual acuity 20/50 or worse improve at least three lines on a Snellen letter chart within 6 months after the onset of symptoms. Optic atrophy is frequently found after the acute episode resolves.
Temporary worsening of vision with physical activity was originally described by Uhthoff in 1890. Uhthoff phenomenon now refers to new or worsening neurologic symptoms that occur in some patients with elevations in temperature (often during exercise or a hot shower). Symptoms are transient and attributed to reversible conduction block along demyelinated nerve fibers.
Oculomotor abnormalities are common. Diplopia usually results from a sixth nerve palsy or internuclear ophthalmoplegia. Other forms of nystagmus (e.g., gaze-evoked or pendular) are also common. Smooth pursuit and saccadic eye movement abnormalities may be observed. Third or fourth nerve palsies, the one-and-a-half syndrome (reflecting damage to the parapontine reticular formation or the sixth nerve nucleus causing ipsilateral gaze palsy as well as impaired adduction in contralateral gaze due to damage to the medial longitudinal fasciculus), opsoclonus, and symptomatic homonymous field defects are rare.
Facial numbness and vertigo can be difficult to distinguish from peripheral vestibular dysfunction, or facial weakness that is usually of the upper motor neuron type, but may mimic an idiopathic Bell palsy. Sudden hearing loss is uncommon and should raise the possibility of Susac syndrome. Intractable hiccups are rare but may result from lesions in the medulla or upper cervical spine. Intractable hiccups or vomiting should raise the possibility of NMO. Dysphagia is uncommon except in advanced MS. Limb weakness is common in MS and may occur during an acute exacerbation or incomplete recovery from an acute attack. The legs are more often affected than the arms and hands.
Approximately 70% of patients have some spasticity, which most commonly involves the legs. Spasticity is often accompanied by painful spasms and upper motor neuron signs. Spasticity often impairs mobility and activities of daily living and disrupts sleep. Gait abnormalities are common and usually a result of ataxia, weakness, or spasticity. However, in some instances, spasticity of the leg extensors permits weight bearing and improves gait. In nonambulatory patients, spasticity may interfere with transfers and personal hygiene.
An extensor plantar response is seldom the only evidence of corticospinal tract dysfunction. Demyelinating lesions in the anterior horn cells and dorsal root entry zones occasionally result in atrophy and hyporeflexia.
Paresthesias, dysesthesias, hypesthesia, or the Lhermitte sign (whereby neck flexion generates an electric sensation that usually radiates from the back of the neck to the lower back and possibly into one or more limbs) are common initial manifestations of MS and occur in most patients at some time.
An action tremor of the arms is common in MS patients. It is often accompanied by other signs of cerebellar disease, including gait ataxia, dysmetria, dysdiadochokinesia, and dysarthria. Tremor of the head, trunk, and legs is much less frequent. Incapacitating tremor, scanning speech, and truncal ataxia are seen in advanced disease.
Paroxysmal symptoms in MS are brief, repetitive, stereotyped attacks of neurologic dysfunction that are thought to result from ephaptic spread of abnormal electrical discharges from partially demyelinated nerve fibers (“cross-talk”). These discharges can arise from areas with acute inflammation and demyelination or chronic tissue damage. The symptoms usually last a few seconds to a few minutes and may occur anywhere from a few to 200 times per day. Attacks may occur spontaneously or be provoked by sudden noises, emotion, movement, hyperventilation, or tactile stimulation. The most common symptoms are trigeminal neuralgia and tonic spasms. Paroxysmal dysarthria, ataxia, diplopia, itching, paresthesias, pain, hemifacial spasm, and glossopharyngeal neuralgia are less frequent.
Trigeminal neuralgia occurs in about 2% of MS patients. The character and quality of the pain is usually indistinguishable from idiopathic trigeminal neuralgia. However, trigeminal neuralgia in MS is more likely to involve both sides of the face and is often accompanied by trigeminal neuropathy or other signs of brain stem dysfunction.
Paroxysmal dystonia, or tonic spasms, the second most common movement disorder in MS (only tremor occurs more frequently), are stereotyped, sometimes painful attacks of unilateral dystonic posturing of the limbs. They are occasionally bilateral and rarely involve the face. The attacks last between 30 seconds and 2 minutes and can occur up to 60 times a day. Tonic spasms are probably caused by demyelinating lesions involving the corticospinal tract. However, they are not pathognomonic for MS and are occasionally seen in patients with cerebral ischemia or spinal cord trauma.
Pseudobulbar affect is characterized by episodes of laughing or crying that do not coincide with the individual’s emotional state. This can cause significant emotional distress to both patients and caregivers.
Fatigue is one of the most common and disabling MS symptoms. It is out of proportion to physical activities and is typically worse in the afternoon. Fatigue may precede the first clinical demyelinating event by months or years in one-third of patients. The etiology of MS-related fatigue is poorly understood. No association exists between fatigue and disease course; disability; brain volume; or MRI lesion load, location, or activity.
Bladder dysfunction, which can result in failure to store or empty urine, or a combination of the two, affects approximately 75% of patients. In 15%, symptoms are severe enough to prevent the patients from leaving home or attending social activities. Demyelinating lesions above the level of the pons may result in detrusor hyperreflexia with uninhibited bladder contractions, which causes urinary urgency that is often accompanied by frequency, nocturia, and urge incontinence. Lesions involving the reticulospinal pathways above S2 and below the pons may also lead to involuntary bladder contractions or cause simultaneous contraction of the bladder wall and urethra, a condition known as detrusor-sphincter dyssynergia. Patients with detrusor-sphincter dyssynergia have storage and emptying dysfunction and a combination of urgency, frequency, difficulty initiating voiding, incomplete emptying, and incontinence. Damage to the upper urinary tract and kidneys as a result of increased intravesicular pressure is rare. Hypocontractility and failure of the bladder to empty properly occurs with demyelination of the lower sacral anterior horn cells. Complete inability to void is uncommon.
Bowel dysfunction often coexists with bladder dysfunction and is present in up to 70% of patients. Constipation, which may be caused by immobility, decreased fluid intake, or medication side effects (especially those used to treat bladder dysfunction), is common.
Sexual dysfunction is present in up to 90% of patients. It is most likely to occur in progressive MS but affects up to two-thirds of patients with relapsing-remitting disease. The most common symptoms among men are erectile and ejaculatory dysfunction. Women have difficulties achieving orgasm, decreased vaginal lubrication, and reduced vaginal sensation. Both men and women may have reduced libido. Physiologic (e.g., fatigue, weakness, spasticity, pain, hypesthesia) and psychological factors (e.g., depression, anxiety about bladder and bowel dysfunction) may interfere with sexual activity. Many of the medications commonly used to treat other MS symptoms, including anticholinergics, antidepressants, and baclofen, can adversely affect sexual function.
Depression is clearly the most common affective disorder in MS with a lifetime prevalence of 50% before age 60 years. Some patients experience depressive symptoms but do not meet diagnostic criteria for major depression. The etiology of depression is likely multifactorial with psychosocial and biologic factors contributing to such a high frequency. There is concern that the β-interferons may cause or exacerbate depression. Episodes of major depression may occur during periods of exacerbations, remission, or disease progression. Although much less common than depression, bipolar disorder, anxiety, pseudobulbar affect, and euphoria are also prevalent in patients with MS.
Cognitive impairment occurs in up to 65% of patients with MS. Short-term memory, attention, concentration, verbal intelligence, visuospatial skills, and information processing are the domains most commonly affected. Disturbances of language and immediate and long-term memory occur less frequently. Disability and disease duration are generally poor predictors of cognitive dysfunction, which can occur early in the course of MS. However, cognitively impaired patients have more lesions, more severe tissue
damage, and smaller brain volumes than unimpaired patients as measured by conventional and nonconventional MRI techniques. Changes detected by magnetization transfer imaging in normalappearing brain tissue may have a stronger correlation with cognitive dysfunction than T1 or T2 lesion load.
In most patients, cognitive dysfunction is subtle and goes undetected in the neurologist’s office. The Mini-Mental State Examination is not useful because it may fail to identify nearly 75% of MS patients deemed to be cognitively impaired by detailed neuropsychological testing.
Approximately 70% of patients experience pain at some time. Pain can occur during an acute exacerbation (e.g., ocular pain with optic neuritis or dysesthesias with demyelinating plaques involving spinal cord), but almost half of MS patients have chronic pain. It is not a major problem for most, but 20% have severe pain. In addition to painful muscle spasms, paroxysmal phenomena, and the sensory symptoms discussed earlier, MS patients are more likely to have joint, muscular, and extremity pain than age- and sexmatched controls. Many patients also complain of neck and back pain, which may be due to posture or gait abnormalities.
Disease Course
No biologic markers or MRI features distinguish the several forms of MS. The current classification system is based on consensus and relies on the clinical course (
Fig. 69.1). Appropriate classification of patients is imperative for appropriate treatment with the disease-modifying drugs (see the following text).
Relapsing-remitting MS is the initial form of the disease in 85% to 90% of patients. It is characterized by acute relapses interspersed with clinical remissions. Symptoms from a relapse typically evolve over several days to a week before reaching a nadir. Recovery is variable, but approximately 40% of relapses result in persistent neurologic deficit and patients may accumulate disability in a stepwise fashion. When left untreated, most patients with relapsing-remitting MS ultimately enter the secondary progressive phase of MS, which is characterized by gradual deterioration with or without occasional superimposed relapses.
Ten percent to 15% of patients have primary progressive MS, which is characterized by continuous and usually gradual deterioration of neurologic function (e.g., a slowly progressive monoparesis). There may be plateaus and slight fluctuations, but relapses do not occur. Fifteen percent to 40% of patients who start with the primary progressive form later have an acute relapse, which may not occur for many years after the original symptoms. This type of MS is referred to as progressive relapsing multiple sclerosis and is the least common form.
Different mechanisms may be responsible for symptomatic worsening. An inflammatory component may result in relapses, whereas neurodegeneration may be responsible for progressive disease.
Pregnancy
Pregnancy is protective in MS, particularly during the third trimester when there is a 70% reduction in the annualized relapse rate compared with the year prior to pregnancy. However, when MS is left untreated, about 30% of women have a relapse in the first 3 months postpartum before the risk returns to the prepregnancy rate 4 to 6 months after delivery. These effects may be the result of changes in Th1 and Th2 immune responses mediated by estriol or vitamin D, both of which are markedly elevated during the final trimester of pregnancy and fall abruptly after delivery; other hormonal changes that occur during pregnancy may also play a role.
Women with relapses during pregnancy or in the year prior to pregnancy are more likely to have a relapse in the first 3 months postpartum than women without relapses during these periods, but it is impossible to accurately predict who will have a relapse. In an uncontrolled study, intravenous immunoglobulin (IVIG) ameliorated the increased risk of postpartum exacerbations. However, the results of a subsequent randomized, double-blind trial were not as encouraging. Furthermore, there is now good evidence from well-designed, large, placebo-controlled trials that IVIG is an ineffective therapy for relapsing-remitting and secondary progressive MS. Breastfeeding does not affect the postpartum course of MS.
Vaccination
Infections, including minor upper respiratory infections, increase the risk of MS exacerbations. Strategies that minimize the risk of infection should be incorporated. Concerns that vaccinations may trigger MS exacerbations are based on anecdotes. However, a prospective, randomized, double-blind, placebo-controlled trial, showed that influenza immunization does not increase the risk of relapse or disease progression and patients should be routinely vaccinated. Hepatitis B, tetanus, and varicella vaccines appear to be safe in MS patients. There are few data regarding the safety of other vaccines in MS, but patients who meet the CDC guidelines should be advised to have them. Patients treated with fingolimod or mitoxantrone should not receive live attenuated vaccines, as there is some risk of developing the infection the vaccine is meant to prevent, or they may have an
attenuated response to the vaccination. It is probably best to delay immunizations for 4 to 6 weeks in the setting of an MS relapse because the goal of relapse management is to diminish the immune system’s reactivity, and vaccination has the opposite effect.