INTRODUCTION
Multiple system atrophy (MSA) is a rare and presumably sporadic neurodegenerative disorder usually occurring during the 6th decade and clinically characterized by a combination of parkinsonism, autonomic, cerebellar, or pyramidal signs. MSA is a progressively incapacitating disorder with a median survival between 6 and 10 years (1). The disease is pathologically defined by diffuse cell loss, gliosis, and abundant α-synuclein–positive oligodendroglial cytoplasmic inclusions (GCIs) (2). MSA is thus cytopathologically defined as a synucleinopathy along with idiopathic Parkinson’s disease (IPD) and dementia with Lewy bodies (DLB) (3). The term “multiple system atrophy” has been coined by Graham and Oppenheimer in 1969 (4) to lump together diseases previously termed “olivopontocerebellar atrophy,” “nigrostriatal degeneration,” and “Shy–Drager syndrome”. It is accepted, since the founder paper of Niall Quinn in 1989, that these “entities” are overlapping forms of expression of the same pathologic entity that is MSA (5). The term “MSA–parkinsonism” (MSA-P) is used for patients with predominant parkinsonism and MSA-cerebellar (MSA-C) for those with predominant cerebellar features, both sharing autonomic failure (AF) as a core clinical feature (1).
EPIDEMIOLOGY
MSA represents 5% to 10% of all causes of primary degenerative parkinsonism with a prevalence estimated somewhere between 2 and 5/100,000 (6–8), the incidence being less well known because of its rarity, probably around 1/100,000/year (8). MSA is usually diagnosed in the sixth decade (mean age of 56.2 ± 8.4 years in the largest European cohort) (9). The disease is mainly sporadic, while no environmental factors or toxins have yet been identified increasing the risk without ambiguity (6). MSA occurs worldwide, while the proportion of MSA-P compared to MSA-C is greater in western countries and that of MSA-C greater in Asian populations. The origin of this difference is unknown, although genetic factors may be involved.
PATHOLOGY AND CYTOPATHOLOGY
The pathology of MSA predominates in nigrostriatal and olivopontocerebellar systems; the relative severity determines the clinical presentation: MSA-P or MSA-C; and in autonomic structures of the medulla and spinal cord in both forms. Usually, in typical and advanced cases the brain appears macroscopically atrophied with brownish shrinkage of the striatum, pale mesencephalon with atrophy of pons, middle cerebellar peduncles, and vermis (10,11). Loss of neurons and myelin fibers as well as reactive gliosis predominate in both systems as well as anatomically and functionally linked regions such as substantia nigra pars compacta and medium spiny striatal neurons on one hand; pontine neurones, Purkinje cells, and olivary complex neurons on the other. Degeneration is also found in the dorsal nucleus of the vagus nerve, the ventrolateral medulla, the pontomedullary reticular formation, the Bötzinger nucleus, the intermediolateral horns, and Onuf’s nucleus of the sacral cord, in relation with AF.
GCIs are the pathologic hallmark of the disease. These inclusions are argyrophilic and half-moon, oval, or conical in shape and are composed of 20- to 30-nm multilayered tubular filaments (Fig. 15.1) (2,3). Neuronal intracytoplasmic and intranuclear inclusions can also be found. Their distribution selectively involves basal ganglia, supplementary and primary motor cortex, reticular formation, and the pontocerebellar system (2). GCIs contain classical cytoskeletal antigens, including ubiquitin and tau, chaperone proteins, and α-synuclein (3). GCIs are distributed throughout the brain, but their density predominates in mostly affected areas such as corticosubcortical motor loops and olivopontocerebellar systems. However, some discrepancy between the density of GCIs and neuronal loss may occur in certain areas.
PATHOPHYSIOLOGY
The most recent hypothesis suggests that GCI represent the primary change in MSA, and the neuronal pathology develops secondary to the glial pathology (12,13) through yet-unclear mechanisms, some of which being cystatin C signaling and p25/TBBP accumulation (14). Usually, mature oligodendrocytes do not express α-synuclein; thus α-synuclein mRNA expression and protein accumulation of insoluble aggregates into filamentous inclusions appear to play a key role in MSA (15). Also, α-synuclein impairs oligodendrocyte maturation (16). In MSA, α-synuclein is abnormally phosphorylated (particularly on serine 129), which may promote fibril formation. Several posttranslational modifications are also found such as abnormal glycosylation and nitration. α-Synuclein accompanying chaperone proteins such as synphilin or 14-3-3 protein may play also a role in inclusions’ pathogenesis (3). Recent data suggest the possibility of transmission of α-synuclein pathology and degeneration in transgenic mice in a prion-like manner such as in IPD (17). α-Synuclein may also be transmitted from neurons to oligodendrocytes. Mitochondrial dysfunction, ubiquitine proteasome system failure, oxidative stress, and neuroinflammation also act as general common pathways leading to neuronal cell death (11,13).
Recently, the contribution of genetic factors was suggested to play a role in MSA, although familial cases are scarce particularly in western countries (18). Genome-wide association studies (GWAS) have suggested a link to α-synuclein variants, but these results were not confirmed in an ongoing study, including 1,000 patients (19,20). More recently, variants and mutations in certain families in the COQ2 gene have been discovered particularly in patients with Asian background (21). This may point to associated mechanism of energy failure along with the synucleinopathy. However, these findings were not replicated in cohorts from Europe, North America, Korea and China.
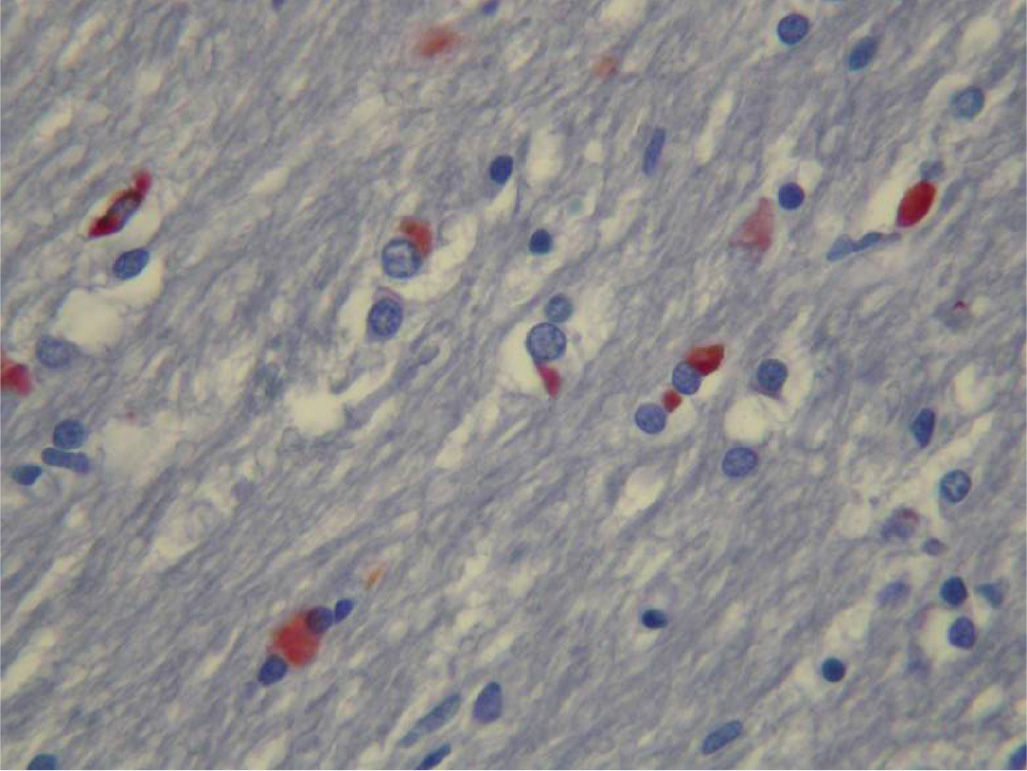
Figure 15.1. Oligodendroglial cytoplasmic inclusions (GCIs) in the substantia nigra, labeled for α-synuclein (in red).
CLINICAL SIGNS AND SYMPTOMS
MSA usually occurs in the sixth decade (mean age at onset is 58 years), while it is relatively accepted that disease onset before 30 and after 80 years is unlikely. In most instances, MSA patients seek medical advice because of motor signs, either parkinsonism in MSA-P or cerebellar signs in MSA-C (9). Patients often present unrecognized prodromal symptoms of AF such as urinary problems, erectile dysfunction, or orthostatic hypotension (OH). Sleep disturbances such as REM sleep behavior disorder (RBD) may also occur decades before the diagnosis (22).
PARKINSONISM
Parkinsonism is present in about 90% of patients at one time of their disease history (23). The classical presentation of parkinsonism in MSA is said “atypical” because several signs and symptoms also referred as “red flags” (Table 15.1) are not expected in typical IPD (24). The most characteristic and frequent presentation is bilateral often symmetric akineto-rigid parkinsonism with early postural instability and falls that is poorly responsive or unresponsive to levodopa. Although typical pill-rolling rest tremor is rare (<10%), any type of tremor, particularly, jerky, irregular, myoclonic, action, and intention tremor is found in two-thirds of patients. Some patients may present with parkinsonism undistinguishable from IPD during the first years, with good to excellent response to levodopa. These patients have been termed as “minimal change MSA” according to limited putaminal pathology at autopsy (10,25).
CEREBELLAR SYNDROME
While it is the presenting and predominant symptom in MSA-C, it is inconstant (40%) and late in MSA-P. Cerebellar signs and symptoms are often difficult to recognize in bradykinetic patients with impaired gait and posture. However, gait ataxia at tandem gait plus one other sign among kinetic ataxia at finger-to-nose and heel-knee-shin tests, cerebellar dysarthria, and oculomotor signs, particularly gaze nystagmus (along with saccadic pursuit, dysmetric saccades, square wave jerks, and abolition of vestibule–ocular reflex by fixation), are the most discriminant to qualify for additional cerebellar syndrome (1).
| Red Flags (24) |
Red flag | Definition |
Early instability with recurrent falls | Within 3 y of disease onset |
Rapid progression | “Wheelchair sign”: dependent <10 y from disease onset |
Orofacial dystonia | Based on clinical judgment |
Camptocormia | Prolonged episodes of forward trunk flexion |
Pisa syndrome | Prolonged episodes of lateral trunk flexion |
Disproportionate antecollis | Severe neck flexion, minor flexion elsewhere |
Contractures of hands or feet | Excluding Dupuytren’s or contracture due to other known cause |
Jerky tremor | Irregular postural or action tremor of the hands and/or fingers with definite myoclonus |
Diurnal inspiratory stridor | Based on clinical judgment |
Nocturnal inspiratory stridor | Based on clinical judgment |
Inspiratory sighs | Involuntary deep inspiratory sighs/gasps |
Severe dysphonia | Based on clinical judgment |
Severe dysarthria | Based on clinical judgment |
Severe dysphagia | Based on clinical judgment |
Random eye movement sleep behavior disorder | Intermittent loss of muscle atonia and appearance of elaborate motor activity associated with dream mentation |
Sleep apnea | Prolonged arrests of breathing |
Excessive snoring | Increase from premorbid level, or newly arising |
Cold hands/feet | New development of coldness and color change of extremities, with blanching on pressure and poor circulatory return |
Raynaud’s phenomenon | New emergence of painful “white fingers” |
Emotional incontinence—crying/laughing | Inappropriate crying without sadness or laughing without mirth |
Past history of documented hypertension | Based on clinical judgment |
AUTONOMIC FAILURE
AF is the signature of MSA, and following consensus criteria AF is a mandatory criterion for the diagnosis of MSA (1). Indeed, AF affects almost all patients early during the disease course contrary to Parkinson’s disease (PD), progressive supranuclear palsy (PSP) and corticobasal degeneration, where it is inconstant, less severe, and occurs later. AF is the presenting symptom in about a third of patients. AF core symptoms are OH and genitourinary impairment. Other symptoms include constipation, gastroparesis, vasomotor dysfunction (cold extremities, cold dusky hands), sweating problems, and nighttime respiratory disorders (26).
Orthostatic Hypotension
OH manifests after postural changes, principally in the morning, after long standing still, after meals, exercise, and under warm atmosphere. Classical symptoms are dizziness, blurred vision, blunted cognition, intense fatigue, occipital headache, and back and buttocks ache. Syncope is rarer but characteristic of the disease. OH may be asymptomatic. OH is defined by a drop of 20 mm Hg of systolic and 10 mm Hg of diastolic blood pressure (BP) within 3 minutes of standing (1). A drop of at least 30 mm Hg systolic and 15 mm Hg diastolic BP is more discriminant for central neurogenic OH and mandatory to qualify for a diagnosis of probable MSA (1). A substantial proportion of patients presents delayed OH after 3 minutes of standing, a time lag which is clinically useful to take into account.
Genitourinary Impairment
Almost universal in MSA, it often precedes other AF symptoms. Erectile dysfunction precedes urinary symptoms in two-thirds of male patients and affects all in later disease stages. Urinary symptoms are bladder instability with frequency, urgency, and incontinence but also urinary retention along with sphincter weakness or bladder sphincter dyssynergia. Retention can be found by ultrasound even in absence of dysuria and may be early in the disease (22,26). Anal incontinence is rare and late in the disease course.
OTHER SIGNS AND SYMPTOMS
Pyramidal Signs
Corticospinal signs are poorly specific and found in about half of patients. They consist in brisk reflexes along with at least one Babinski sign. A motor deficit or a spastic paraparesis is not accepted as a manifestation of MSA (1).
Other Movement Disorders
About a third of patients have postural, stimulus-sensitive, or action-triggered cortical myoclonus in the distal parts of the upper limbs (27). Spontaneous (not drug-induced) dystonia is frequently observed in MSA (about half of patients) and often gives rise to fixed dystonic postures: foot and hand dystonia, trunk dystonia such as antecollis, or Pisa syndrome. Facial dystonia may also be observed as a side effect of levodopa (28).
Swallowing Problems
Accompanying mixed hypophonic and quivering irregular voice, swallowing problems are often observed already early in the disease. They carry the risk of aspiration pneumonia, a frequent cause of death in MSA (29). Excessive salivation and drooling is frequent and a consequence of reduced automatic swallowing.
Pain
At least half of patients report pain which is rather similar than that found in PD in nature: bone and joint pain, dystonic pain, neuropathic-like pain (30).
Breathing Disorders
Breathing disorders may be the first symptom of AF (31). Deep sighs, dyspnea, irregular or periodic Cheyne–Stokes-like respiration, stridor, and sleep apnea syndrome are the most frequent manifestations. Stridor, a paradoxical closing of vocal cords during inspiration, is the most severe and distressing disorder occurring in 13% up to 69% of patients (32). It first occurs at night but can later also manifest during daytime. Stridor is due to vocal cord paresis and/or dystonia and is associated with a poorer survival and increased risk of sudden death.
Sleep Problems
Along with nighttime stridor, central or obstructive apneas are very frequent, affecting up to two-thirds of patients (33). Bed partners should be carefully interviewed and educated about the different sound pitch of snoring, sleep apneas, and stridor, and the potentially harmful consequences of RBD. Sleep apneas and altered breathing rhythm during sleep may increase the risk of nighttime sudden death which is the most frequent cause of death in MSA along with aspiration pneumonia. RBD are observed in almost all patients with MSA at one time of their disease and may precede the disease onset by more than 10 years (34). Daytime sleepiness and fatigue are very frequent complaints of MSA patients.
Mood, Behavior, and Cognition
Depression is very prevalent particularly in MSA-P and constitutes one of the main cause of altered quality of life (35,36). Emotional and mood swings with inappropriate laughing or crying can also be observed, particularly in patients with severe cerebellopontine involvement in MSA-C. Suicide thoughts and attempts seem more prevalent in MSA-P than in PD.
Although early dementia is not within the clinical spectrum of MSA, cognitive deficits and late dementia are increasingly recognized. Dysexecutive patterns of cognitive alteration are similar in nature but with a severity ranging between that observed in PD and PSP (37). Different patterns of cognitive deficits seem to exist between MSA-P and MSA-C (37). Dementia occurs in at least 10% of MSA patients, a probably underestimated feature due to the difficulties in diagnosing dementia in severely disabled patients and applicability of conventional criteria.
NATURAL HISTORY AND DIAGNOSIS CRITERIA
Diagnosis of MSA remains difficult, and the mean delay between onset and diagnosis varies from 2 to 7 years (9). The first set of diagnosis criteria have been proposed in 1989 by Niall Quinn (5), then refined and extended by consensual diagnosis criteria in 1998 which were revised in 2008 (7). Three levels of certainty are proposed and displayed in Table 15.2—“possible,” “probable,” and “certain”—if pathological diagnosis is obtained. The most difficult differential diagnosis remains MSA-P versus IPD (38). PSP patients without prominent oculomotor signs (PSP-P) who have urinary problems may also be confounded with MSA-P. Patients with Lewy body dementia (DLB) presenting with marked AF along with parkinsonism and mild dementia may also be difficult to differentiate from MSA. MSA-C may be confused with other cerebellopontine degenerative disorders, particularly spinocerebellar ataxia and fragile X-associated tremor/ataxia syndrome (39).
| Diagnosis Criteria (1) |
CRITERIA FOR THE DIAGNOSIS OF PROBABLE MULTIPLE SYSTEM ATROPHY (MSA)
A sporadic, progressive, adult (>30 y)-onset disease characterized by
1. Autonomic failure involving urinary incontinence (inability to control the release of urine from the bladder, with erectile dysfunction in males) or an orthostatic decrease of blood pressure within 3 min of standing by at least 30 mm Hg systolic or 15 mm Hg diastolic and
2. Poor levodopa-responsive parkinsonism (bradykinesia with rigidity, tremor, or postural instability) or
3. A cerebellar syndrome (gait ataxia with cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction)
CRITERIA FOR THE DIAGNOSIS OF POSSIBLE MSA
A sporadic, progressive, adult (>30 y)-onset disease characterized by
1. Parkinsonism (bradykinesia with rigidity, tremor, or postural instability) or
2. A cerebellar syndrome (gait ataxia with cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction) and
3. At least one feature suggesting autonomic dysfunction (otherwise-unexplained urinary urgency, frequency or incomplete bladder emptying, erectile dysfunction in males, or significant orthostatic blood pressure decline that does not meet the level required in probable MSA) and
4. At least one of the following features:
• Possible MSA–parkinsonism (MSA-P) or MSA-cerebellar (MSA-C)
• Babinski sign with hyperreflexia
• Stridor
• Possible MSA-P
• Rapidly progressive parkinsonism
• Poor response to levodopa
• Postural instability within 3 y of motor onset
• Gait ataxia with cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• Dysphagia within 5 y of motor onset
• Atrophy on MRI of putamen, brain stem, or cerebellar peduncle, pons, or cerebellum
• Hypometabolism on FDG-PET in putamen, brain stem, or cerebellum
• Possible MSA-C
• Parkinsonism (bradykinesia and rigidity)
• Atrophy on MRI of putamen, brain stem, or cerebellar peduncle, pons
• Hypometabolism on FDG-PET in putamen
• Presynaptic nigrostriatal dopaminergic denervation on SPECT or PET




