Myasthenia gravis (MG) is caused by a defect of neuromuscular transmission (NMT) owing to antibody-mediated attack on nicotinic acetylcholine receptors (AChR) or muscle-specific tyrosine kinase (MuSK) at neuromuscular junction. It is characterized by fluctuating weakness that is improved by inhibitors of cholinesterase.
EPIDEMIOLOGY
Greater awareness and improved diagnosis has led to an increased incidence of MG. A systematic review spanning 1980 to 2007 of 31 studies mostly conducted in Europe found incidence rates from 3 to 30 per million per year. The prevalence in the United States is estimated at 20 per 100,000 by Philips in 2003. MG is equally present in men and women older than the age of 40 years; however, it is three times more common in women than men under 40 years of age. MuSK myasthenia is also more common in women.
Systemic autoimmune disorders are found with greater incidence in patients with MG. Thyroid disorders are present in up to 15% of patients with MG. Other autoimmune disorders with increased incidence in patients with MG include rheumatoid arthritis and systemic lupus erythematosus (SLE).
Familial cases are rare; single members of pairs of fraternal twins and several sets of identical twins have been affected. Young women with MG tend to have HLA-B8, HLA-DR3, and HLA-DQB1* 0102 haplotypes; in young Japanese women, HLA-A12 is prominent. These observations imply the presence of a linked immune response gene that encodes a protein involved in the autoimmune response. First-degree relatives show an increased incidence of other autoimmune diseases (SLE, rheumatoid arthritis, thyroid disease) and HLA-B8 haplotype.
PATHOBIOLOGY
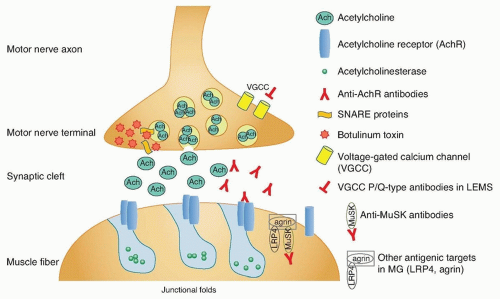
The purpose of the neuromuscular junction is to transmit the electrical impulse from the motor neuron to the muscle fiber and trigger contraction. The complex synapse is depicted in
Figure 89.1. An electrical signal reaching the terminal motor axon results in
the release of acetylcholine (ACh) quanta into the synaptic cleft through fusion of vesicles with the membrane. The released ACh serves as a chemical signal, which then travels across the synaptic cleft and binds to AChR on the postsynaptic muscle fiber membrane, triggering a muscle fiber action potential and subsequent contraction or twitch.
The neuromuscular junction is susceptible to autoimmune pathology because there is no blood-nerve barrier to protect the neuromuscular junction. The precise mechanisms triggering autoimmunity at the neuromuscular junction are unknown; however, there is considerable knowledge regarding the pathobiology. MG is an antibody-mediated disorder in which the autoantibodies are clearly pathogenic. This is deduced from several lines of evidence:
Identification of circulating serum antibodies against the AChR and other components of the neuromuscular junction
Immunization of experimental animal models with purified AChR induces high titers of anti-AChR antibodies, weakness, and pathologic and electrophysiologic features of MG.
Passive transfer of human immunoglobulin G (IgG) from persons with MG to mice reproduces electrophysiologic features of MG.
Plasma exchange reduces anti-AChR antibody titers and ameliorates MG signs and symptoms.
Extensive research has shown the complex autoimmune nature of MG. Antibodies against the AChR are found in 80% to 85% of patients with MG. A majority of these are AChR “binding” antibodies. AChR “modulating” antibodies are found in less than 1% of MG patients without binding antibodies, and “blocking” antibodies are found in approximately 10% of MG patients without binding antibodies. The polyclonal IgG antibodies to AChR are produced by plasma cells in peripheral lymphoid organs, bone marrow, and thymus. These cells are derived from B cells that have been activated by antigen-specific T-helper (CD41) cells. The T cells have also been activated, in this case, by binding to AChR antigenic peptide sequences (epitopes) that rest on the histocompatibility antigens on the surface of antigen-presenting cells.
Most common are anti-AChR binding antibodies, which bind to the AChR on the postsynaptic membrane and cause the complement-mediated destruction of the junctional folds and accelerate internalization and degradation of AChR. Blocking antibodies prevent ACh binding to the AChR. Modulating antibodies distort the neuromuscular junction and folds. The end result in all cases is a loss of functional AChR. The AChR antibodies react with multiple determinants, and enough antibodies circulate to saturate up to 80% of all AChR sites on muscle. A small percentage of the anti-AChR molecules interfere directly with the binding of ACh, but the major damage to endplates seems to result from actual loss of receptors owing to complement-mediated lysis of the membrane and to acceleration of normal degradative processes (internalization, endocytosis, lysosomal hydrolysis) with inadequate replacement by new synthesis. As a consequence of the loss of AChR and the erosion and simplification of the endplates, the amplitude of miniature endplate potentials is about 20% of normal, and patients are abnormally sensitive to the competitive antagonist curare. The characteristic decremental response to repetitive stimulation of the motor nerve reflects failure of endplate potentials to reach threshold so that progressively fewer fibers respond to arrival of a nerve impulse. Most AChR antibodies are directed against antigenic determinants on the extracellular portion of the protein farthest out from the membrane rather than the ACh-binding site. The summed effects of the polyclonal anti-AChR antibodies, especially those that fix complement, result in destruction of the receptors. Physiologic studies indicate impaired postsynaptic responsiveness to ACh, which accounts for the physiologic abnormalities, clinical symptoms, and beneficial effects of drugs that inhibit acetylcholinesterase (AChE). If binding or blocking antibodies are absent, tests can be done for “modulating” antibodies that enhance degradation of the receptors in cultured cells, bringing the number of positive tests to 90%, according to Howard et al.
The MuSK protein is localized to the inner surface of the muscle membrane and is involved in AChR clustering. Approximately 10% of all patients with MG and 50% of patients without antibodies to the AChR will have antibodies against MuSK. There is prominent atrophy of muscles in anti-MuSK MG. Anti-MuSK antibodies are pathogenic by inhibiting the clustering of the AChR, demonstrated in passive transfer of human IgG from patients with anti-MuSK MG to rodents.
Beginning in 2011, multiple groups of researchers found other antigenic targets in seronegative MG patients. Lipoproteinrelated protein 4 (LRP4) is essential for neuromuscular junction formation, serving as an agrin receptor and participating in agrinactivated MuSK and AChR clustering. Antibodies against LRP4 were found in MG patients seronegative for AChR antibodies and MuSK antibodies. Antibodies against agrin were identified in patients both with anti-AChR antibodies and in seronegative patients. There are likely additional autoantibodies to components of the neuromuscular junction which are yet to be discovered and account for the remainder of the “seronegative” MG patients.
How the autoimmune disorder starts is not known. In human disease, in contrast to experimental MG in animals, the thymus gland is almost always abnormal; often multiple lymphoid follicles show germinal centers (“hyperplasia of the thymus”), and in about 15% of patients, there is an encapsulated benign tumor, a thymoma. These abnormalities are impressive because the normal thymus is responsible for the maturation of T cells that mediate immune protection without promoting autoimmune responses. AChR antibodies are synthesized by B cells in cultures of hyperplastic thymus gland. The hyperplastic glands contain all the elements needed for antibody production: class II HLA-positive antigen-presenting cells, T-helper cells, B cells, and AChR antigen; that is, mRNA for subunits of AChR has been detected in thymus, and “myoid cells” are found in both normal and hyperplastic thymus. The myoid cells bear surface AChR and contain other muscle proteins. When human myasthenic thymus was transplanted into severely congenitally immunodeficient mice, the animals produced antibodies to AChR that bound to their own motor endplates, even though weakness was not evident.
T lymphocytes are responsible for initiating and maintaining the autoantibody response, and the thymus gland is the site of overt pathology. About 70% of thymus glands from adult patients with MG are not involuted, weigh more than normal, and demonstrate lymphoid hyperplasia. In contrast, germinal centers in non-myasthenics are numerous in the lymph nodes and spleen but are sparse in the thymus. Immunocytochemical methods indicate that the thymic germinal centers in myasthenics contain B cells, plasma cells, HLA class II DR-positive T cells, and dendritic cells. Approximately 10% of patients with MG will have lymphoepithelial thymomas, and the neoplastic thymic epithelial cells express self-antigens including the AChR and other skeletal muscle proteins such as titin and the ryanodine receptor. The lymphoid cells in these tumors are T cells; the neoplastic elements are epithelial cells. Benign thymomas may nearly replace the gland with only residual glandular material at the edges, or they may rest within a large hyperplastic gland. Thymomas tend to occur in older patients, but in one series, 15% were found in patients between ages 20 and 29 years. They may invade contiguous
pleura, pericardium, or blood vessels or seed onto more distant thoracic structures, including the diaphragm; however, they almost never spread to other organs. In older patients without thymoma, the thymus gland appears involuted, often showing hyperplastic foci within fatty tissue on microscopic examination of multiple samples. Excessive and inappropriately prolonged synthesis of thymic hormones that normally promote differentiation of T-helper cells may contribute to the autoimmune response. Still another possible initiating factor is immunogenic alteration of the antigen AChR at endplates; penicillamine therapy in patients with rheumatoid arthritis may initiate a syndrome that is indistinguishable from MG except that it subsides when administration of the drug is stopped.
There are few familial cases of the acquired autoimmune disease, but disproportionate frequency of some HLA haplotypes (B8, DR3, DQB1) in MG patients suggests that genetic predisposition may be important. Other autoimmune diseases also seem to occur with increased frequency in patients with MG, especially hyperthyroidism and other thyroid disorders, SLE, rheumatoid arthritis, pernicious anemia, and pemphigus.
In about 50% of cases, muscles contain lymphorrhages, which are focal clusters of lymphocytes near small necrotic foci without perivascular predilection. In a few cases, especially in patients with thymoma, there is diffuse muscle fiber necrosis with infiltration of inflammatory cells; similar lesions are only rarely encountered in the myocardium. Lymphorrhages are not seen near damaged neuromuscular junctions (although inflammatory cells may be seen in necrotic endplates in rat experimental autoimmune MG), but morphometric studies have shown loss of synaptic folds and widened clefts. Some nerve terminals are smaller than normal, and multiple small terminals are applied to the elongated, simplified postsynaptic membrane; others are absent. Other endplates appear normal. On residual synaptic folds, immunocytochemical methods show Y-shaped antibody-like structures, IgG, complement components 2 and 9, and complement membrane attack complex.
CLINICAL FEATURES
The symptoms of MG have three general characteristics that together provide a diagnostic combination (
Table 89.1). Formal diagnosis depends on clinical features, demonstration of the response to cholinergic drugs, electrophysiologic evidence of abnormal neuromuscular transmission, and demonstration of circulating antibodies to AChR or MuSK.
The fluctuating nature of myasthenic weakness is unlike any other disease. The weakness varies in the course of a single day, sometimes within minutes, and it varies from day to day or over longer periods. Major prolonged variations are termed remissions or exacerbations; when an exacerbation involves respiratory muscles to a degree that intubation and assisted mechanical ventilation, it is called a crisis. An “exacerbation” may be ameliorated by noninvasive ventilation or may be a transient worsening without respiratory distress. Variations sometimes seem related to exercise; this and the nature of the physiologic abnormality have long been termed excessive fatigability, but there are practical reasons to de-emphasize fatigability as a central characteristic of MG. Patients with the disease less often complain of fatigue or symptoms that might be construed as fatigue except when there is incipient respiratory muscle weakness. Myasthenic symptoms are always owing to weakness and not to rapid tiring. Fatigue is common in MG, and at times can be difficult (especially for patients) to differentiate from true myasthenic exacerbation.
The second characteristic of MG is the distribution of weakness. Ocular muscles are affected first in about 40% of patients and are ultimately involved in about 85%. Ptosis and diplopia are the symptoms that result. Other common symptoms affect facial or oropharyngeal muscles, resulting in dysarthria, dysphagia, and limitation of facial movements. Together, oropharyngeal and ocular weakness cause symptoms in virtually all patients with acquired MG. Limb and neck weakness is also common in conjunction with cranial weakness. Almost never are the limbs affected alone. MuSK myasthenia particularly involves ocular, facial, and bulbar muscles and has high risk of respiratory insufficiency. MuSK myasthenia can follow a rapidly progressive and more severe course.
Aside from the fluctuating nature of the weakness, MG is not a steadily progressive disease. The general nature of the disease, however, is usually established within weeks or months after the first symptoms. If myasthenia is restricted to ocular muscles for 2 years, certainly if it is restricted after 3 years, it is likely to remain restricted, and only in rare cases does it then become generalized. Distinguishing solely ocular myasthenia from generalized myasthenia soon after onset is challenging.
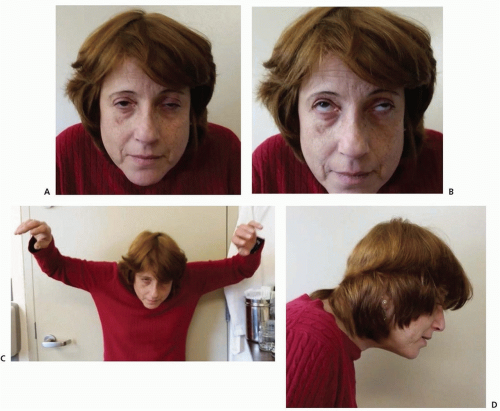
Vital signs and findings on general physical examination are usually normal, unless the patient is in crisis. Weakness of the facial and levator palpebrae muscles produces a characteristic expressionless appearance with drooping eyelids, which can be asymmetric (
Fig. 89.2A). Weakness of ocular muscles (
Fig. 89.2B) may cause paralysis or weakness of isolated ocular muscles, limitation of conjugate gaze, complete ophthalmoplegia in one or both eyes, or a pattern resembling internuclear ophthalmoplegia. Proximal limb weakness is manifest by difficulty in raising the arms above the head (
Fig. 89.2C) and difficulty climbing stairs, whereas weakness of the neck flexor and extensor muscles may result in head drop (
Fig. 89.2D). Weakness of oropharyngeal or limb muscles, when present, can be shown by appropriate tests. Respiratory muscle weakness can be detected by pulmonary function tests, which should not be limited to measurement of vital capacity but should also include inspiratory and expiratory pressures, the measurement of which may be abnormal even before overt symptoms are evident. Muscular wasting of variable degree is found in about 10% of patients but is not focal and
is usually encountered only in patients with malnutrition caused by severe dysphagia. Fasciculations do not occur, unless the patient has received excessive amounts of cholinergic drugs. Sensation is normal and the reflexes are preserved, even in muscles that are weak.
Crisis is most likely to occur in patients with oropharyngeal or respiratory muscle weakness. It seems to be provoked by respiratory infection in many patients or by surgical procedures, including thymectomy, although it may occur with no apparent provocation. Both emotional stress and systemic illness may aggravate myasthenic weakness for reasons that are not clear; in patients with oropharyngeal weakness, aspiration of secretions may occlude lung passages to cause rather abrupt onset of respiratory difficulty. Major surgery may be followed by respiratory weakness without aspiration, however, so this cannot be the entire explanation.