and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Endocrine and metabolic disturbances are probably the most common causes of neuropathy (Table 17.1). In these situations, the diagnosis is based on clinical and laboratory findings, examination of peripheral nerve biopsy having no role in management. Nevertheless, the pathologist should be aware of the spectrum of pathology seen in each entity, as biopsy may be performed in such patients on the suspicion that another pathological process is at play. Toxic neuropathies other than alcohol are considered separately in Chap. 18.
Table 17.1
Endocrine/metabolic neuropathies
Endocrine |
Diabetes |
Hypothyroidism (?hyperthyroidism) |
Acromegaly |
Organ disease |
Uremic neuropathy |
Neuropathy in liver disease |
Nerve alterations in pulmonary disease |
Vitamin deficiencies |
Vitamin B1 (thiamine) |
Vitamin B6 (pyridoxine) |
Vitamin B12 (cobalamin) |
Vitamin E (α-tocopherol) |
Folate |
17.1 Diabetic Neuropathy
The global prevalence of diabetes is predicted to increase from 366 million in 2011 to 552 million in 2030 (Whiting et al. 2011) and will likely lead to a marked increase in diabetic neuropathy, which is currently the single most common neuropathy in the world and a leading cause of morbidity in this disease. One frequently quoted prospective study of 4,400 patients documented clinically evident neuropathy in 7.5 % of diabetics at the time of initial diagnosis, increasing linearly with time to approximately 50 % at 25 years (Pirart 1978a, b). More recent data indicates that although over half of diabetics have a neuropathy of some sort, it is most often subclinical, and significant symptomatology is seen in only a small fraction (Dyck et al. 1993). Neuropathy develops in both type 1 and type 2 diabetes, is associated with retinopathy and nephropathy, and increases in frequency and severity with duration of diabetes, age, hypertension, smoking (in people with type 1 diabetes), patient height, alcohol consumption, and worsening degree of glycemic control (Llewelyn et al. 2005).
17.1.1 Clinical Manifestations
Diabetic neuropathy is not a single neuropathy; rather, it is a complex spectrum of neuropathies (Table 17.2), including symmetrical, asymmetric (involving cranial and somatic nerves), and autonomic neuropathies (Thomas and Tomlinson 1993; Llewelyn et al. 2005). The first group includes sensory or sensorimotor polyneuropathy, autonomic neuropathy, and symmetrical proximal motor neuropathy. The second group includes cranial neuropathies, trunk and limb mononeuropathies, and asymmetric lower limb motor neuropathy. A number of patients may present very early with painful small fiber neuropathy (detectable with skin biopsy but not as frequently with sural nerve biopsy) at the time they only exhibit an impaired glucose tolerance test (Smith et al. 2001; Novella et al. 2001).
Table 17.2
Classification of diabetic neuropathies
Impaired glucose tolerance and hyperglycemic neuropathy |
Generalized (symmetrical) neuropathies |
Sensorimotor |
Acute painful (including treatment induced) |
Autonomic (parasympathetic, sympathetic, enteric, visceral sensory) |
Acute motor (unusual) |
Asymmetric (focal and multifocal neuropathies) |
Cranial nerves (III, IV, VI) |
Thoracolumbar |
Upper limb |
Lumbosacral radiculoplexus (Bruns–Garland syndrome) |
Superimposed chronic inflammatory demyelinating neuropathy (CIDP) |
Hypoglycemic neuropathy |
17.1.1.1 Distal Symmetrical Sensorimotor Neuropathy
Symmetrical length-dependent and predominantly sensory neuropathy is the most common, manifesting with numbness, variable severity of pain and paresthesias, impaired vibration sense, and loss of distal reflexes. When sensory loss extends above the knees, it develops in the fingers, spreading to the hands and forearms as it progresses proximally in the lower limbs. The anterior aspect of the trunk can become affected due to the involvement of the distal territory of the intercostal nerves and crown of the head due to the involvement of the longest fibers of the trigeminal nerve (Sabin et al. 1978). Distal motor manifestations can be associated but are rarely dominant. Although usually mild, at its most extreme form, this sensorimotor polyneuropathy may cause a sensory ataxic, acrodystrophic, or arthropathic picture and culminate in amputation. Small fiber neuropathy manifesting with pain, hyperesthesia, and autonomic disturbance can be present in isolation, or associated with the sensorimotor syndrome. Although symptoms usually evolve slowly, an acute painful diabetic neuropathy can appear in the setting of poor diabetic control and weight loss (Asbury et al. 1963), or conversely with initiation of insulin treatment (Llewelyn et al. 1986). Diabetic neuropathy can also present with pain, proximal weakness and tissue wasting, and minimal sensory deficits, evolving insidiously and symmetrically, or acutely and focally (Barohn et al. 1991; Bastron and Thomas 1981). The simultaneous presence of several types of neuropathy is common.
In symmetrical sensorimotor neuropathy, electrophysiological studies reveal a loss of distal amplitudes, especially in sensory fibers. Conduction slowing is common, although rarely to the extent usually present in demyelinating neuropathies such as CMT-1 or CIDP (Behse et al. 1977; Thomas and Tomlinson 1993). EMG in the asymmetric diabetic nerve syndromes reveals multifocal regions of denervation, with involvement of paraspinal muscles often suggesting a nerve root lesion (Bastron and Thomas 1981).
A growing body of data suggests that severity of peripheral nerve manifestations correlates with severity of hyperglycemia over a long-term period and that optimal glycemic control results in amelioration or at least delayed progression of diabetic neuropathies (Committee Health Care Issues ANA 1986; DCCT Research Group 1993). At one time the use of aldose reductase inhibitors in treatment and prevention of diabetic neuropathy held great promise, but clinical results have been largely disappointing (Harati 1992; Tsai and Burnakis 1993).
17.1.1.2 Asymmetric Diabetic Neuropathies
This category (recently reviewed by Younger 2011; Prasnoor et al. 2013) includes diabetic lumbosacral radiculoplexus neuropathy (DLRPN; also known as “diabetic amyotrophy,” “femoral neuropathy of diabetes,” and “proximal diabetic neuropathy” or “Bruns–Garland syndrome”), cranial nerve palsies (usually cranial nerves III, IV, and VI), truncal radiculopathy, and upper limb mononeuropathy. Early in diabetes, occasional middle-aged and elderly men with type 2 diabetes, often following institution of insulin therapy or associated with significant weight loss, experience sudden or subacute onset of (typically unilateral) motor dysfunction with weakness and muscle wasting, involving large nerves to the lower limbs or cranial nerves. Although the condition usually begins in one leg, spread to the other leg may occur within weeks or months, and, in a third of the cases, proximal arm muscle involvement (cervicobrachial radiculoplexopathy) has been reported. Occasionally, upper limb involvement occurs. Oculomotor nerve palsies are the most common cranial neuropathy observed in diabetic patients which occurs in diabetic patients over 50 years of age, both in insulin- and in noninsulin-dependent diabetic patients. Spontaneous complete recovery invariably occurs within 2–3 months.
17.1.1.3 Diabetic Autonomic Neuropathy
Both sympathetic and parasympathetic neuropathy occur in diabetes resulting in a myriad of autonomic complaints, which may present individually or in groups (Rundles 1945) involving cardiovascular, genitourinary, alimentary, and sudomotor functions. The presence of symptomatic diabetic autonomic neuropathy significantly increases morbidity and mortality (Ewing et al. 1980).
17.1.2 Pathology
Discussion of histological findings is complicated by the co-occurrence of several diabetic neuropathy variants which may have different pathogenic mechanisms. Biopsy findings in diabetic neuropathy are invariably nonspecific, and this invasive procedure should be performed for diagnostic purposes only if a treatable alternative is being considered.
17.1.2.1 Pathology in Diabetic Symmetrical Sensorimotor Neuropathy
Light Microscopy
Sensorimotor diabetic neuropathy is a predominantly axonal process (Behse and Buchthal 1977; Dyck et al. 1986a, b; Johnson et al. 1986; Yagihashi and Matsunaga 1979; Schmidt and Bilbao (in press)) (Fig. 17.1a, b). Behse and co-workers (1977) observed that in predominantly sensory neuropathies, large and small myelinated fibers were equally depleted, while in sensorimotor neuropathy the large myelinated fibers (MFs) were more severely affected (Behse and Buchthal 1977). However, Dyck et al. (1986b) found no evidence of selective depletion of small or large MFs. Regenerating clusters can be prominent, but with severe neuropathy they often diminish in number. Although the neuropathy can be very chronic, actively degenerating axons may be seen, and the overall appearance is not as indolent as that of neuronal Charcot–Marie–Tooth disease (CMT). Nonetheless, studies of “early” diabetic neuropathy have reported normal myelinated fiber density, axonal area, and G-ratio in the presence of teased fiber studies showing paranodal abnormalities, segmental demyelination, and remyelination without active degeneration of myelinated axons.
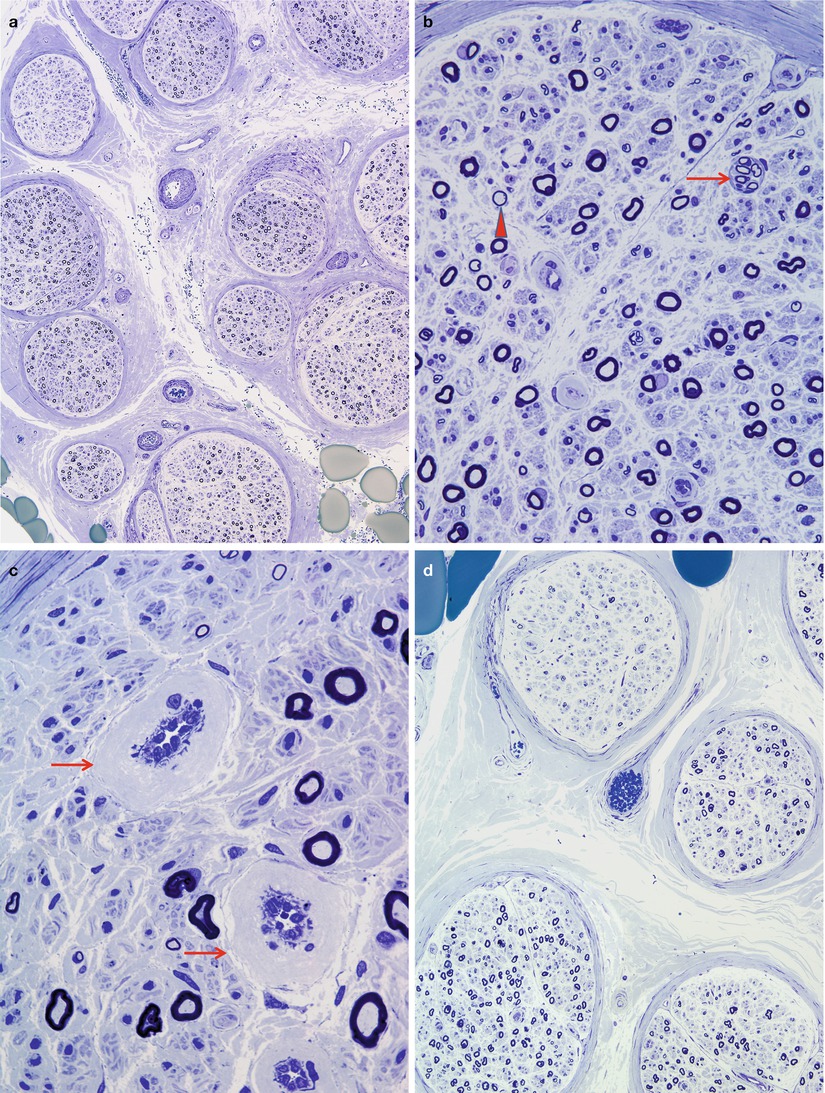
Fig. 17.1
Diabetic neuropathy: (a, b) moderate reduction in myelinated fiber density, regenerating clusters (arrow, b) disproportionately thin myelin sheaths (arrowhead, b) and marked thickening of vessel walls (arrows, c). There may be significant fascicle-to-fascicle variability in axon number (d) (1 μm plastic sections. Magnifications: a, 100×; b, 400×; c, 1,000×; d, 200×)
We have been impressed by the frequency of focal nerve lesions in these patients, often appearing as perineurial-based regions where myelinated fibers are reduced in number, actively degenerating, or thinly myelinated (Figs. 17.2, 17.3, and 17.4). The perineurium at such sites may appear disrupted, and rarely a few randomly directed myelinated fibers may escape the confines of the nerve fascicle, forming a miniature “traumatic” neuroma (Figs. 17.2 and 17.4) which appears most common in nerves of diabetics. It is intuitive to regard such abnormalities as nerve “infarcts” although this implies an ischemic pathogenesis (vide infra). The epineurial vasculature is typically less severely involved, although vasculopathy has been reported.
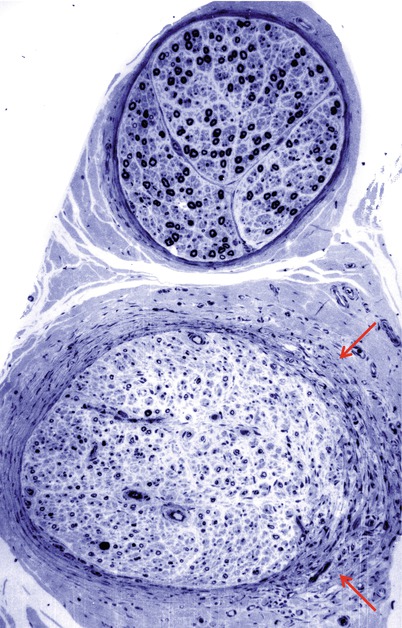
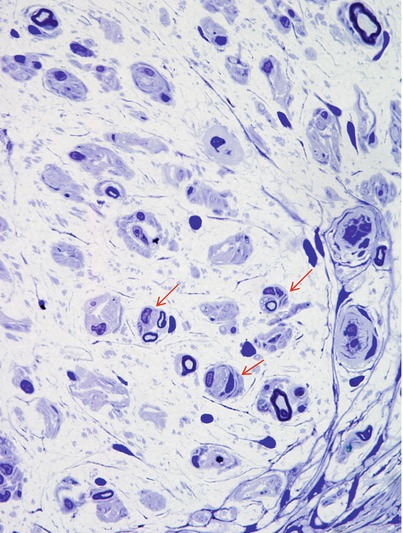
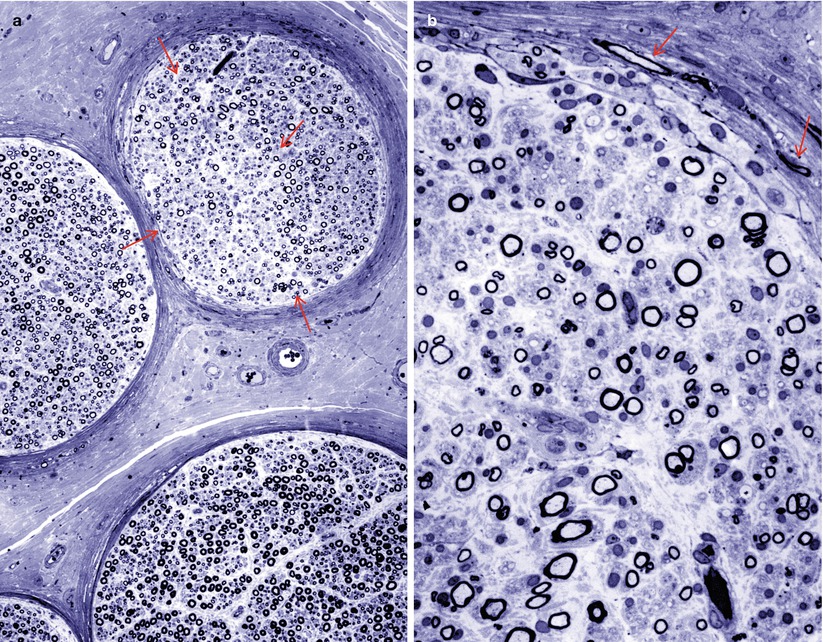
Fig. 17.2
Diabetic neuropathy, reparative phase following focal fascicular damage. Compare MF density between two adjacent fascicles. Note perineurial damage (arrows) (1 μm plastic sections, 200×)
Fig. 17.3
Diabetic neuropathy: high-power view of affected fascicle in Fig. 17.2 showing pseudo-onion bulbs (arrows) resulting from axonal regeneration through a region of focal damage (1 μm plastic sections, magnification, 600×)
Fig. 17.4
Diabetic neuropathy, reparative phase following focal fascicular damage. Subtle fascicular damage is shown characterized by a geographic region of diminished numbers fibers with attenuated myelin (outlined by arrows, a). Note aberrant regeneration of fibers in perineurium (arrows, b) (1 μm plastic sections, magnification a, 200×; b, 600×)
Sima and colleagues have suggested a difference in the pattern of axonal pathology depending on patient age and type of diabetes: younger patients with IDDM are less likely to display multifocal nerve degeneration, and the typical axonal alteration on teased fibers is myelin wrinkling, while older patients with NIDDM demonstrate multifocal nerve lesions, with Wallerian degeneration on teased fibers (Sima et al. 1988a). In support of this observation, Llewelyn et al. (1988) found no difference in the nonuniformity of fiber loss between a group of relatively young diabetics and in control nerve taken from patients with CMT-1.
Segmental myelin changes usually take the form of inappropriately thinly myelinated axons indicating remyelination or regeneration, with cross sections revealing ongoing demyelination considerably less frequently (Figs. 17.1, 17.5, and 17.6b). Teased fibers demonstrate the spectrum of myelin alteration from paranodal widening to demyelinated segments. In asymptomatic diabetics, segmental myelin alterations may precede loss of axons (Chopra et al. 1969; Vital et al. 1974). Onion-bulb (OB) formations can be seen in diabetic neuropathy but have probably been overemphasized (Fig. 17.7). The OBs are most often rudimentary and do not dominate the histological picture as with CMT-1 or some cases of CIDP (Ballin and Thomas 1968). Indeed, onion-bulb-like structures (pseudo-onion bulbs) are more likely to be a reflection of axonal regeneration (Fig. 17.7a, b). Thomas has commented that in retrospect, the prominent onion-bulb formations described in a case of “diabetic” neuropathy in his 1966 report (Thomas and Lascelles 1966) were likely due to concurrent CIDP (Thomas and Tomlinson 1993). Some authors have reported that the myelin alterations appeared to be independent of axonal changes (Behse and Buchthal 1977), while others have emphasized clustering along certain fibers indicating secondary demyelination (Dyck et al. 1986b).
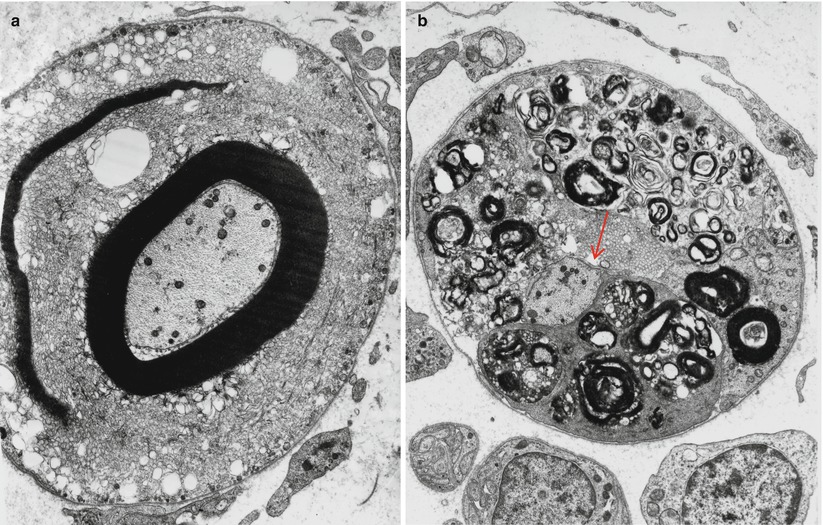
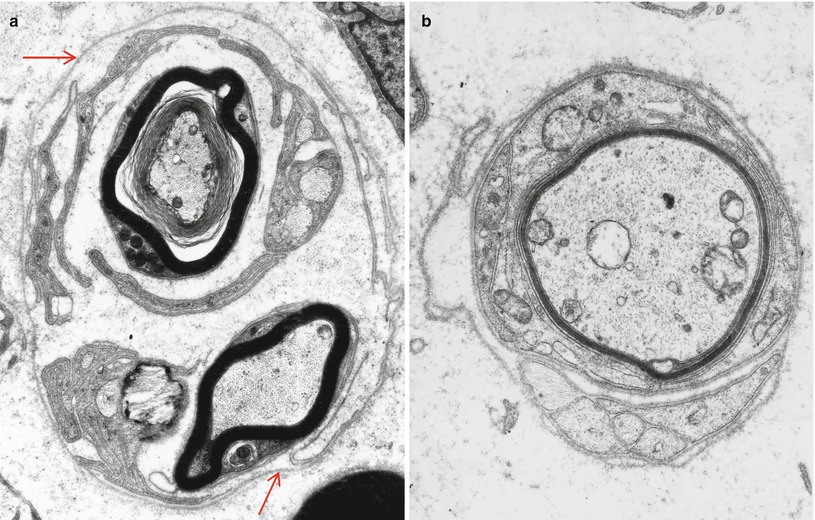
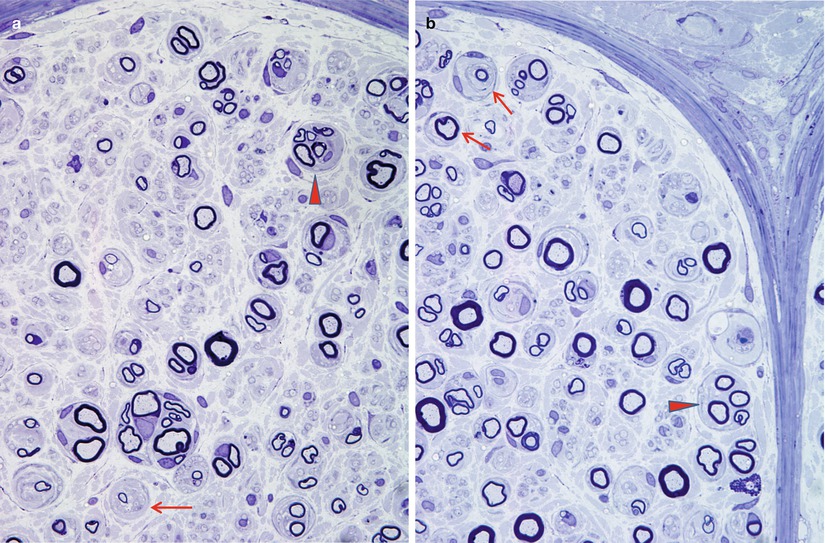
Fig. 17.5
Diabetic neuropathy: segmental demyelination. Note vesicular pattern in (a). The demyelinated axon (arrow, b) is surrounded by Schwann cells containing myelin debris (a ×8,892, b ×6,384)
Fig. 17.6
Diabetic neuropathy, regenerative processes: in (a) note continuous basement membrane (arrows) surrounding a regenerating cluster of myelinated and unmyelinated fibers. Remyelination is illustrated in (b) (a ×9,576, b ×15,048)
Fig. 17.7
Diabetic neuropathy: note axon loss with (pseudo)-onion bulbs (arrows, a, b) and regenerating cluster (arrowheads, a, b) (1 μm plastic sections. Magnification: a, b, 1,000×)
Hyalinization of endoneurial microvessels is a typical feature of diabetic nerves, regardless of the presence or absence of neuropathy (Fig. 17.1c). This may reach striking proportions, but these microvascular changes remain a nonspecific finding seen in many chronic neuropathies. Alternatively, the severity of endoneurial microvascular alterations has been correlated with the severity of the neuropathy (Malik et al. 1993). Endoneurial area may be increased (Behse and Buchthal 1977), with expansion of the endoneurial and subperineurial extracellular matrix (Ballin and Thomas 1968). Changes in epineurial microvessels are usually mild by comparison (Malik et al. 1993). A low-grade mononuclear inflammatory infiltrate is infrequently seen.
Electron Microscopy
The ultrastructural correlate of vascular hyalinization is reduplication and thickening of microvascular basement membranes (Fig. 17.8a, b). Other alterations which have been emphasized in diabetic neuropathy include endothelial cell hypertrophy and hyperplasia, reduction in capillary luminal area, and microvessel “closure” (Dyck et al. 1985, 1986b; Malik et al. 1989) (Fig. 17.8c, d). However, not all workers have confirmed these findings, and they may be seen in other chronic neuropathies (Bradley et al. 1990) or be related to age (Sima et al. 1988a). Obstruction of capillary lumina with degenerate cellular material and fibrin has been described (Timperley et al. 1985; Yasuda and Dyck 1987), but is uncommon in our experience (Fig. 17.8d, e).
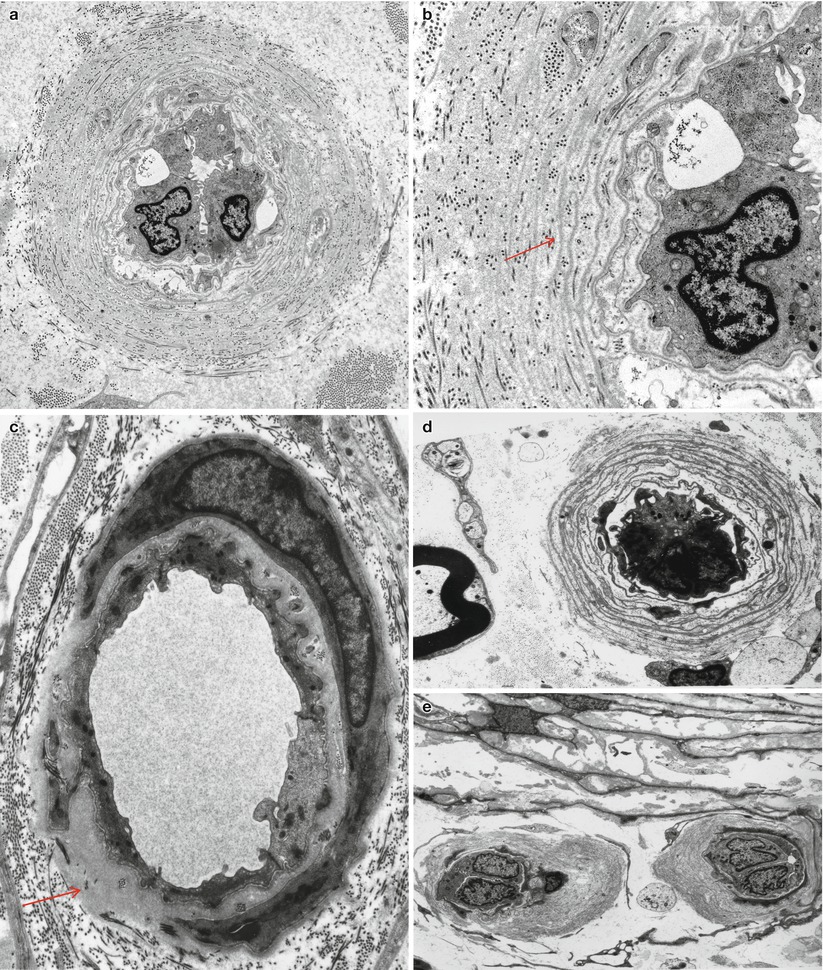
Fig. 17.8
Diabetic neuropathy: the range of endoneurial microvascular alterations is illustrated. Marked duplication of basal lamina about endothelium and pericytes (a, b). In (c) there is thickening of endothelial basal lamina (arrow, c). “Closed” vessels are illustrated in (d, e) (a, 6,000×; b, 15,000×; c, 8,379×; d, 5,426×; e, 2,880×)
The full range of nonspecific axonal alterations may be seen. One lesion which has received considerable attention is “axoglial dysjunction,” referring to ultrastructural and molecular findings resulting in the disruption of the characteristic junctional complexes joining terminal myelin loops to the axolemma at the paranodal area (Sima 1993; Sima et al. 2000). Swelling of the paranodal axon is often associated with this finding. Axoglial dysjunction seems to be more significant in type 1 (younger) patients than in type 2 (Sima et al. 1988a), but its specificity is uncertain (Sladky et al. 1991). Other differences between types 1 and 2 diabetes in the neuropathologic findings of diabetic somatic nerves have also been described in animals and humans. Type 1 diabetics are reported to develop fewer foci of patchy axonal degeneration and more evidence of myelinopathy than those with type 2 diabetes, in which axonal degeneration is most prominent.
Increased numbers of denervated Schwann cell subunits give early evidence of involvement of unmyelinated fibers, and at later stages of the disease, unmyelinated fiber counts can drop below the lower limit of normal. An apparent increase in rigidity and durability of Schwann cell basal lamina has been observed in diabetics, where after axonal degeneration and regeneration the original basement membrane outline is abnormally maintained (King et al. 1989) (Fig. 17.6a). Perineurial cells may display a thickened basal lamina (Johnson et al. 1981; King et al. 1989).
17.1.2.2 Pathology in Painful/Small Fiber Neuropathy (Sural and Skin Biopsy)
In three patients described by Brown and colleagues, nerve biopsy revealed more severe loss of small MFs than of large MFs (Brown et al. 1976). Unmyelinated axons were not severely depleted, but showed a striking shift to the left in size range, with only 4 % more than 1 μm in diameter (control 33 %). The Schwann cell–axon relationships appeared altered in the unmyelinated fibers, with the frequent occurrence of axons incompletely surrounded by Schwann cells or abutting one another. Two reports by Said and colleagues include a total of nine patients with clinical features suggesting small fiber disease (Said et al. 1983a, 1992). A dramatic depletion of unmyelinated fibers was noted, often to 10 % or less of the lower limit of normal. Myelinated fibers were also severely affected, but to a slightly lesser degree. Regenerating clusters were frequent. A proximodistal gradient of severity compatible with distal axonopathy was observed. Segmental myelin abnormalities were seen in a high proportion of teased fibers and thought to occur both primarily and secondary to axonal alterations (Said et al. 1983a, 1992). Onion-bulb and “pseudo”-onion bulbs (formed by regenerating clusters) were noted.
In contrast to the above observations, other workers (Behse and Buchthal 1977; Dyck et al. 1986b; Llewelyn et al. 1991) have failed to identify biopsies showing a selective involvement of small myelinated and unmyelinated axons, even in patients with severe autonomic or painful neuropathies. Such cases may represent the extreme manifestations of a single type of symmetrical diabetic neuropathy (Dyck et al. 1986b; Llewelyn et al. 1991). Nerve biopsy in an acute painful neuropathy associated with establishment of tight glycemic control biopsy revealed only a chronic axonal neuropathy with no special features (Llewelyn et al. 1986). Another group of patients suffering from acute painful neuropathy associated with severe weight loss did not display any special features except perhaps an increased degree of active axonal degeneration (Archer et al. 1983).
Skin biopsies have been recently used in the analysis of diabetic symmetrical sensory polyneuropathy and autonomic neuropathy (illustrated in Chap. 1, Fig. 1.2a, b and Fig. 1.8) (Lauria et al. 2005; Luo et al. 2011; McCarthy et al. 1995; Periquet et al. 1999). Fiber loss demonstrable in skin biopsies has been reported in the absence of axon loss in the sural nerve in diabetic sensory neuropathy, which reflects loss of the most distal sensory complement of axons. The ability to perform multiple skin biopsies in the essential absence of neuropathic residua is one of the assets of skin biopsy and can potentially be used to follow the progress of treatment. After capsaicin-induced sensory skin denervation, diabetic patients show a slower rate of intraepidermal nerve fiber regeneration compared with healthy subjects, a result which may identify incipient neuropathy by regenerative stress at the earliest stages in which it may be most amenable to therapy. The demonstration of distal preterminal nerve fiber swellings in skin biopsies of diabetics may represent a “pre-degenerative” change and relate to synaptic/nerve terminal pathology in DRG and autonomic ganglia. The skin biopsy technique has been expanded to glabrous, non-hairy skin demonstrating in diabetic patients a significant reduction of both mechanoreceptive Meissner corpuscles and their afferent myelinated nerve fibers (Peltier et al. 2013). Myelinated nerve fiber loss was correlated with the decreased amplitudes of sensory/motor responses in nerve conduction studies. Skin biopsies may distinguish neuropathies from radiculopathies, since dorsal roots are proximal to DRG and do not directly produce epidermal nerve changes.
Recent studies (Cheng et al. 2013) have examined skin biopsies from nondiabetic controls, diabetic patients without neuropathy, diabetics with neuropathy and neuropathic pain, and diabetics with neuropathy but without pain. They reported that patients with painful diabetic neuropathy showed a higher percentage of GAP43-immunoreactive epidermal axons compared to total numbers of PGP fibers (suggesting regenerating axons) and increased numbers of tropomyosin-receptor kinase A and substance P containing axonal swellings per PGP-immunoreactive axon, suggesting that the axons are associated with nociception. The DRG of autopsied diabetic patients contain neurofilament-laden, calcitonin gene-related peptide (CGRP)-, trkA-, and substance P-immunoreactive dystrophic swellings involving recurrent collateral axons that terminate on adjacent perikarya (Schmidt et al. 1997) but have no proven association with painful neuropathy. Other levels of the CNS (e.g., integration in the spinal cord or higher brain centers) may also contribute to painful neuropathy.
17.1.2.3 Pathology in Asymmetric Diabetic Neuropathy
The array of names given to the proximal, often asymmetric, diabetic neuropathies betrays the uncertainty that exists regarding the site of the lesion. At times this syndrome has been termed “diabetic amyotrophy,” “Bruns–Garland Syndrome,” “diabetic polyradiculopathy,” “diabetic lumbosacral plexopathy,” or “diabetic femoral neuropathy” (also see Chap. 1, Sects. 1.3, 1.4). The usual coexistence of symmetrical and asymmetric polyneuropathy confounds interpretation of nerve biopsy material. In 10 sural nerve biopsies, myelinated fiber loss was almost invariably seen, with the nerve damage distributed in both multifocal and diffuse patterns (Barohn et al. 1991). This group of neuropathies may involve an autoimmune component resulting in an inflammatory microvasculitis with T-cell infiltrates (Younger et al. 1996; Dyck et al. 1999; Prasnoor et al. 2013), although not typically angionecrosis, involving the epineurial vasculature (Fig. 17.9a, b) with endoneurial and perineurial hemosiderin deposition. The possibility that immune-mediated and inflammatory lesions are important in proximal diabetic neuropathy has been suggested by workers who have observed low-grade perivascular inflammatory infiltrates, sometimes associated with vascular occlusion and focal axonal loss, in this setting (Costigan et al. 1990; Said et al. 1994). Frank necrotizing vasculitis has not been illustrated. Vasculopathy accompanies perineuritis (Fig. 17.9a, b) and an axonopathy, which ranges from scattered degenerating axons to the formation of an “injury neuroma” (Llewelyn et al. 1998; Said et al. 2003). Frequently there is fascicle-to-fascicle variability in axon loss (Fig. 17.9b) At least some “infarcts” described in early studies (Raff et al. 1968) may have represented Renaut bodies (Sugimura and Dyck 1981; Thomas and Tomlinson 1993). Other workers have shown only patchy demyelination or fibrosis in the roots or plexus (Bastron and Thomas 1981). A demyelinating picture suggestive of CIDP was seen in nerve biopsy of three patients with “proximal diabetic neuropathy,” and the patients improved with immune suppressive therapy (Costigan et al. 1990). Loss of intraepidermal nerve fibres has also been described in biopsies taken from symptomatic areas. Diabetic LRPN histological findings are identical to those seen in patients with nondiabetic LRPN, although it has been suggested that “nondiabetic” patients with LRPN may also have abnormal glucose tolerance (Kelkar and Hammer-White 2005). The usual course results in spontaneous recovery over several months.
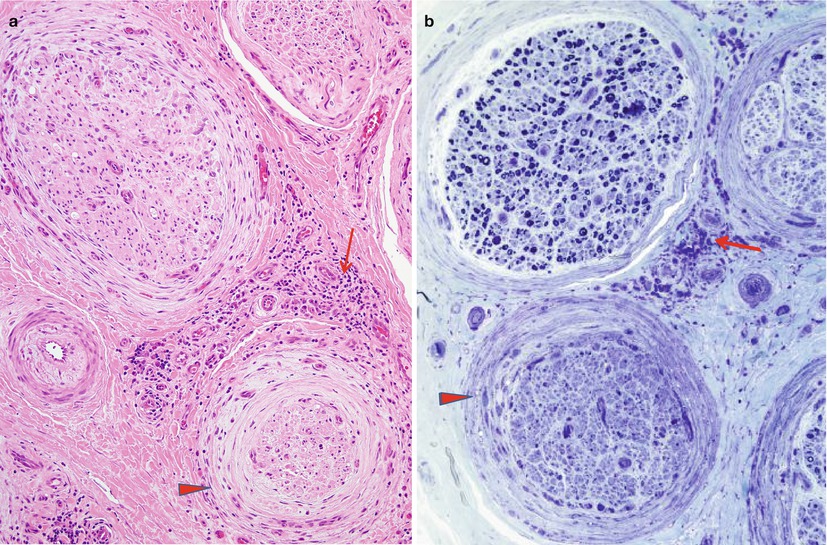
Fig. 17.9
Diabetic lumbosacral radiculoplexus neuropathy (DLPRN): characteristic pathological findings include perivascular inflammation involving the epineurium (arrows, a, b), perineuritis with infiltrating macrophages (arrowheads, a, b), and differential fascicular axon loss (b) (a, H&E paraffin, 200×; b, 1 μm plastic section, 200×)
17.1.2.4 Pathology of Diabetic Autonomic Neuropathy
Investigation of the pathology of diabetic autonomic neuropathy has been neglected in comparison to symmetrical sensorimotor neuropathy (reviewed in Schmidt 2002). Systematic autopsy studies of diabetic patients have demonstrated the development of markedly swollen neurofilament-laden axons and synapses (neuroaxonal dystrophy) in the relative absence of neuron loss in diabetic prevertebral sympathetic autonomic ganglia that serve the bowel and viscera (Schmidt et al. 1993). The presence of lymphocytic infiltrates in some diabetic sympathetic ganglia has been noted, although without proof of an immune pathogenesis. Significant loss of preganglionic myelinated axons and elements of the abdominal vagus nerve has also been described.
The examination of the innervation of sweat glands, arrector pili, and arterioles extends the usefulness of skin biopsies to the analysis of autonomic nerves in diabetic autonomic neuropathy (Kennedy 2004) by concentrating on sweat gland nerve fiber density, neuropathic symptoms, neurological deficits, and sweat production in diabetic patients.
17.1.3 Pathogenesis
17.1.3.1 Site of the Lesion
The length-dependent nature of diabetic symmetrical sensorimotor polyneuropathy and the consistent finding of axonal loss in patients with established neuropathy indicate unequivocally that axonal degeneration is the predominant pathology. Whether the frequently observed myelin alterations occur secondary to axonal influences or are due to a primary schwannopathy remains unsettled. The latter possibility has been favored by the description of segmental myelin changes in patients with early or presymptomatic neuropathy (Chopra et al. 1969; Dyck et al. 1980; Thomas and Lascelles 1966; Vital et al. 1974). No evidence for axonal atrophy was found by Sugimura and Dyck (1981). However, some workers have found remyelinating internodes to cluster along certain axons, suggesting secondary demyelination (Dyck et al. 1986b; Said et al. 1983a, 1992). Said and colleagues proposed that both primary and secondary demyelination occurred in diabetic neuropathy (Said et al. 1983a, 1992).
The fiber types involved in symmetrical diabetic neuropathy may vary between different syndromes. In the most common distal symmetrical sensorimotor polyneuropathy, there is either no selectivity or a preferential involvement of large myelinated fibers. In painful sensory neuropathy with prominent autonomic dysfunction, involvement of small myelinated and unmyelinated fibers may be selective. A predominantly motor neuropathy is rarely observed. It remains unclear if these distinct syndromes are part of the spectrum of the same pathophysiological process or if different pathogenic mechanisms are at play.
17.1.3.2 Cause of the Lesion
The precise pathogenesis of diabetic neuropathy remains unknown (Harati 1992; Thomas 1992). Nonetheless, a number of pathogenetic mechanisms have been proposed and tested in experimental animal models (Table 17.3) which have been recently reviewed (Llewelyn et al. 2005; Obrosova 2009; Vincent et al. 2011). There may be no single mechanism underlying diabetic neuropathy, rather multiple mechanisms may participate and may vary between different forms of diabetic neuropathy. Tight glucose control does delay the development of somatic and autonomic neuropathy, but it does not prevent it, even intensive diabetic treatment of type 2 diabetics does not correct established neuropathy (UK Prospective Diabetes Study Group 1998). Two main schools of thought have existed regarding the cause of the neuropathy. The distally predominant and symmetrical pattern, combined with the known metabolic alterations of diabetes, has suggested that metabolic axonopathy with or without schwannopathy directly underlies nerve changes (Sima 1993). Conversely, the focal nature of some diabetic neuropathies has led to consideration of ischemic causes, and such hypotheses have gradually been extended to the entire spectrum of diabetic neuropathies (Dyck 1989).
Table 17.3
Diabetic neuropathy – proposed pathogenetic mechanisms
Mechanism | Synopsis | References |
|---|---|---|
Oxidative stress | Increased production of oxidant stressors and/or reduction in antioxidant defenses. DRG neurons from diabetic rats show low MnSOD and increased reactive oxygen species associated with dystrophic axonal swellings and impaired axon outgrowth which may lead to axonal degeneration | |
Mitochondriopathy | Insulin- and NT-3-sensitive decrease in mitochondrial membrane potential | |
Nutrient excess interferes with activity of AMP kinase and PGC-1α in sensing neuronal metabolic demands in diabetic DRG, decreasing mitochondrial respiratory chain and Krebs cycle function which may interfere with peripheral nerve energy supply and result in distal axonopathy, a result reversible with resveratrol | ||
Calcium homeostasis | Axons of diabetic DRG neurons exhibit aberrant Ca2+ homeostasis resulting from suboptimal neuronal SERCA activity | Zherebitskaya et al. (2011) |
Neurotrophic substances | Deficiencies in the amount or regulation of various neurotrophic substances (e.g., NGF, NT-3, IGF-I, insulin, erythropoietin, C-peptide) | |
Polyol pathway | Excess glucose is converted to sorbitol and then to fructose using aldose reductase and sorbitol dehydrogenase, diminishing glutathione and increasing oxidative stress. | Obrosova (2002) |
Ischemia | (See text) | |
Advanced glycation end products (AGEs) | Hyperglycemia-induced glycosylation of various intracellular and extracellular proteins converted into AGEs which cross-link a variety of proteins and subcellular organelles and increase oxidative stress by binding to AGE receptors (RAGEs) | |
Mitogen-activated protein kinases | Mitogen-activated protein kinases (MAPKs) are activated by glucose, and oxidative stress may induce excessive protein (e.g., neurofilament) phosphorylation | Purves et al. (2001) |
Neuronal apoptosis | A contentious mechanism, proposed from in vitro cell culture studies conflicting with largely maintained numbers of dorsal root and sympathetic ganglia in experimental rat models, even in the face of increased neuronal activated caspase 3. Mice with experimental diabetes apparently do lose sensory neurons and axons | |
Protein kinase C | Increased activity of protein kinase C (PKC) βII isoform may injure the microvasculature and induction of TGF-β effects on the extracellular matrix | Way et al. (2001) |
Axonal transport | Axonal transport defects have been identified in diabetic animal models involving a variety of materials | Sidenius and Jakobsen (1987) |
Abnormal axonal regeneration/synaptic turnover | Abnormal regeneration and synaptic turnover have been proposed to explain dystrophic nerve terminals and synapses in diabetic DRG and prevertebral sympathetic ganglia | |
Autoimmune pathogenesis | Autoimmune attack has been proposed to explain diabetic autonomic neuropathy in which a variety of antibodies against parasympathetic and sympathetic ganglia have been found, although unproven as a cause of clinical diabetic autonomic neuropathy | |
Neuronal insulin resistance | Neuronal insulin resistance is thought to alter PI3K/pAkt/GSK-3β signaling pathways, mitochondrial biogenesis and function, oxidative stress, and 12/15 lipoxygenase activation | Kim and Feldman (2012) |
Dyslipidemia | Dyslipidemia is a major independent risk factor for the development of neuropathy as shown by epidemiologic studies and demonstration of dysregulated genes associated with lipid metabolism in experimental animals | Vincent et al. (2009) |
Channelopathy | Dysregulation of Na+ and K+ channel activities as a consequence of posttranslational modifications | |
ER stress | Unfolded protein response (UPR) is insufficient to alleviate diabetes-induced ER stress resulting in apoptosis | O’Brien et al. (2014) |
The Metabolic Hypothesis
The metabolic hypothesis began in earnest with the analysis of the role of the polyol pathway in the pathogenesis of diabetic neuropathy (Sima et al. 1988a; Sima 1993). Intraneural concentrations of sorbitol and fructose are increased in both experimental and human diabetic neuropathy, presumably as a consequence of increased metabolic activity along the aldose reductase pathway but do not have an osmotic effect on nerve function. The increase in intracellular sorbitol was reported to influence intracellular myoinositol content and producing a subsequent defect in Na+/K+-ATPase activity (Sima et al. 1988a). These biochemical changes were thought to cause accumulation of Na+ at the nodal axon where sodium channels are found, causing node swelling and detachment of the terminal myelin loops which attach to the axon, so-called axoglial dysjunction. This results in widening of the node and exposure of K+ channels which hyperpolarize the axonal membrane and impair conduction. Sima and co-workers have found that aldose reductase inhibitors promote regeneration of peripheral nerve in diabetic neuropathy (Sima et al. 1988b).
Against this theory stands the failure of aldose reductase inhibitors (Tsai and Burnakis 1993) to have a substantial clinical effect in many studies, although controversy continues. Salutary use of aldose reductase inhibitors would reasonably be expected to interrupt the process in its earliest phases, in order to improve diabetic neuropathy.
Recent work reveals that hyperglycemia in diabetes triggers a multitude of biochemical metabolic changes (see Table 17.3) which are currently thought to underlie the development of diabetic neuropathy. Currently, Fernyhough and co-workers (Chowdhury et al. 2010, 2011, 2013; Fernyhough et al. 2003) propose that nutrient excess in neurons mediates mitochondrial damage through alteration of the AMP-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) signaling axis. This signaling pathway is thought to affect an energy-sensing metabolic pathway which modulates mitochondrial function, biogenesis, and regeneration. The bioenergetic phenotype (maximal oxygen consumption rate, coupling efficiency, respiratory control ratio, and spare respiratory capacity) of mitochondria in diabetic neurons is aberrant as the result of changes in expression and activity of respiratory chain components brought about as a direct consequence of abnormal AMPK/PGC-1α signaling. Other investigators have confirmed and expanded these ideas, demonstrating decreased PGC-1α and downregulation of its transcription factors including TFAM and NRF1 in mouse streptozotocin-diabetic dorsal root ganglion neurons (Choi et al. 2014). A neuropathic phenotype is exacerbated in PGC-1α (−/−) diabetic mice accompanied by a severe neuropathy with reduced mitochondrial DNA and a further increase in protein oxidation. Conversely, overexpression of PGC-1α in cultured adult mouse neurons prevents oxidative stress associated with increased glucose levels (Choi et al. 2014). The result of these changes in cellular bioenergetics is a reduced adaptability to fluctuations in ATP demand, thought to be distally accentuated. This diabetes-induced maladaptive process is hypothesized to result in exhaustion of the ATP supply in the distal nerve compartment and induction of axonal degeneration, a result which can be corrected by resveratrol treatment (Chowdhury et al. 2012, 2013).
The Ischemic Hypothesis
Proponents of the ischemic hypothesis have emphasized the primacy of axonal lesions, their multifocal distribution, alterations in endoneurial and epineurial vessels, and association with disease in other organs (eye, kidney) where microvascular injury is thought to play a central role (Dyck et al. 1986a, b; Dyck 1989 ; Johnson et al. 1986; Sugimura and Dyck 1982; Timperley et al. 1985). The observation that endoneurial microvascular changes differ quantitatively and qualitatively from those seen in other tissues in diabetics mitigates against the suggestion that these simply reflect nonspecific systemic microvasculopathy (Malik et al. 1989, 1993). The distally predominant symmetrical pattern of clinical involvement may result from the summation of numerous random focal insults, with the longest fibers sustaining the greatest number of injuries (Johnson et al. 1986; Waxman et al. 1976). In support of this hypothesis, reduced microvascular luminal area, increased numbers of closed endoneurial capillaries (Dyck et al. 1985; Malik et al. 1993), and plugging of vessel lumina by degenerate cellular material have been described (Timperley et al. 1985; Yasuda and Dyck 1987) and debated. The observation of increased resistance of diabetic peripheral nerve to ischemia (Thomas and Tomlinson 1993) may suggest chronic neural ischemia in these patients.
Correlation of the severity of microvasculopathy and neuropathy has supported an ischemic pathogenesis (Malik et al. 1993; Yasuda and Dyck 1987), but similar microvascular alterations are seen in chronic neuropathies in the absence of ischemia (Bradley et al. 1990). Indeed, endothelial cell hyperplasia was more prominent in CMT-1 than in diabetes and reduced vascular luminal area, and capillary “closure” has not been consistently detected (Bradley et al. 1990), or found to be no different than in age-matched controls (Sima et al. 1991). Moreover, the finding of multifocal nerve fiber loss of equal degree in CMT-1 and diabetic neuropathy weakens the assertion that this pattern suggests an ischemic neuropathy (Bradley et al. 1990; Dyck 1989; Llewelyn et al. 1988). Sima and colleagues (Sima et al. 1988a) found multifocal degeneration of axons in NIDDM (mean age 56) but not IDDM (mean age 38) patients with neuropathy, suggesting that multifocality of nerve lesions in diabetes may relate to age. Geographic regions of fibers with attenuated myelin (Fig. 17.4b) may represent foci of axonal degeneration and subsequent regeneration showing the hypomyelination typical of regenerating fibers or may indicate demyelination with remyelination. Johnson and colleagues (1986) found that the frequency of such focal lesions proximally correlated with the degree of distal nerve fiber depletion in patients with symmetrical sensorimotor neuropathy. The multifocality of the axonal lesions was verified statistically by Dyck et al. (1986b). However, multifocal loss of axons is not pathognomonic of vasculopathy or ischemia and is typical in some forms of inherited neuropathy. Endothelial cell and pericyte degeneration, endothelial cell hyperplasia/hypertrophy, decreased capillary luminal area, and altered blood–nerve barrier permeability are seen in endoneurial microvasculature in patients with diabetic symmetrical sensorimotor neuropathy, findings that correlated with electrophysiological measures of the severity of the neuropathy (Watkins and Thomas 1998; Malik et al. 1993; Giannini and Dyck 1995). Nonetheless, Theriault and colleagues (1997) measured nerve blood flow using laser Doppler flowmetry in diabetic patients with early polyneuropathy and determined that local blood flow was not decreased, even in the presence of definitive evidence of decreased sensory nerve action potential (SNAP) amplitudes and decreased numbers of axons. Finally, axon and Schwann cell nodal and paranodal changes similar to those emphasized in diabetes have been produced by chronic endoneurial ischemia alone (Sladky et al. 1991). Alterations in microvessel junctional structures, similar to “axoglial dysjunction,” have been described (Sima et al. 1991). Increased sural nerve water content due to accumulation of osmotically active solutes may cause a rise in endoneurial pressure and result secondarily in endoneurial ischemia (Griffey et al. 1988; Kalichman and Myers 1991). Nonenzymatic glycosylation of intracellular and extracellular structures may directly cause axon dysfunction or cause cellular and extracellular alterations resulting in endoneurial ischemia (Harati 1992; King et al. 1989).
In summary, the role of microvascular pathology in diabetic neuropathy has been variously interpreted as evidence for an ischemic pathogenesis in diabetic neuropathy or as unrelated but parallel changes (Zochodne 2002); however, there is evidence for tissue hypoxia in the nerve and ganglia in experimental animal models and humans (Zochodne and Ho 1992). Thus, nerve ischemia is associated with nerve dysfunction but may not represent the primary or major cause; rather, it may simply accelerate the process (Llewelyn et al. 2005).
Other Mechanisms
The ischemic and metabolic hypotheses are not mutually exclusive and can be unified by suggestion that metabolic alterations in the neural milieu cause direct or indirect injury to the neural blood supply. An overview of the number of hypotheses proposed to underlie development of neuropathy in diabetes is provided in Table 17.3. It is clear that many of the hypotheses proposed may be interdependent (e.g., oxidative stress, mitochondriopathy, polyol pathway, etc.).
17.2 Neuropathy in Thyroid Disease
17.2.1 Clinical Manifestations
Peripheral neuropathy (excluding carpal tunnel syndrome) is seen in 17–60 % of hypothyroid patients (Nickel et al. 1961; Watanakunakorn et al. 1965). Typically this is a mild, predominantly sensory polyneuropathy with prominent paresthesias. Physical examination is confounded by the frequent coexistence of muscle disease. Electrophysiological studies can be suggestive of a demyelinating or an axonal process, but are usually mixed. CSF protein may be increased. Thyroid replacement is efficacious, but recovery is not always complete (Dyck and Lambert 1970; Nemni et al. 1987; Pollard et al. 1982). Nerve biopsy studies are mixed favoring axonal degeneration (Nemni et al. 1987) or demyelination (Dyck and Lambert 1970).
17.2.2 Pathology
Deposition of mucopolysaccharide materials, a characteristic systemic feature of myxedema, was described in endoneurium and subperineurium in an early nerve biopsy study (Nickel et al. 1961), but subsequent reports have not confirmed this (Dyck and Lambert 1970; Meier and Bischoff 1977; Nemni et al. 1987; Pollard et al. 1982; Shirabe et al. 1975). Renaut bodies were seen in only 2 of 17 cases presented by these latter authors. A predominance of segmental demyelination has been noted in some reports (Dyck and Lambert 1970; Shirabe et al. 1975), while, in others, the degeneration of myelinated and unmyelinated axons has been emphasized (Meier and Bischoff 1977; Nemni et al. 1987; Pollard et al. 1982). It is, however, agreed that larger myelinated fibers are most prominently involved. The neuropathy is usually histologically indolent, and regenerating clusters may be readily visualized. Reich Pi granules may be more prominent than usual (Dyck and Lambert 1970; Meier and Bischoff 1977). Onion-bulb formations have rarely been observed.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


