Axon loss in the optic nerve in multiple sclerosis. These images show significant reduction in normal axonal density in the optic nerves of patients with MS using a neurofilament stain (NF). Panel A shows loss of normal fiber bundles at 10x, whereas Panel B show loss of fibers themselves (40x) accompanied by an increased glial response.
Interestingly, significant inflammation was detected in the retinas of these subjects even though the samples were taken at the end of life. Many of the subjects had secondary progressive disease and the principle inflammatory response would be presumed to have abated. The study demonstrated further evidence of retinal inflammation in a subset of cases subjected to additional immunohistochemical assessment. This inflammation was characterized by the appearance of large numbers of cells expressing MHC Class II molecules (defining them as of immune lineage) throughout the retina, some of which were concentrated in the space around blood vessels (Figure 14.2). The rate of frank retinal periphlebitis was similar between the patients with progressive and relapsing MS. Foamy appearing cells that appeared to be of innate immune lineage were also seen, many of which were outside of the normal microglial network surrounding the inner nuclear layer of the retina (Figure 14.3). The finding of infiltrating and migrating native immune cells distributed throughout the retina means that some of the retinal layers seen using standard retinal imaging protocols may be constituted by cells that are not found in the healthy retina [1].
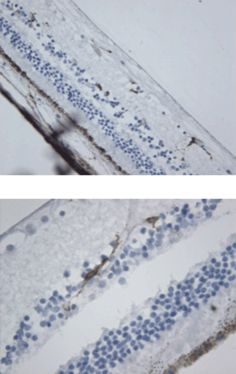
Immune activation in the retina in MS. HLA-DR staining showing MHC class II + cells in the inner retina, including in the retinal nerve fiber, ganglion cell, and inner nuclear/plexiform layers at 20x (Panel A) and inner nuclear layer at 40x (Panel B).
The retinal nerve fiber layer is defined at its inner edge by the internal limiting membrane and at its outer edge by the ganglion cell layer. Although the inner edge is a true biological interface, the outer edge is in large part conceptual. It is defined by the theoretical boundary between the cell soma of retinal ganglion cells and their axonal projections. The boundary appears sharp on optical coherence tomography images in part because of how OCT signal is produced – axonal microtubules reflect light differently when they are perpendicular to the light source.
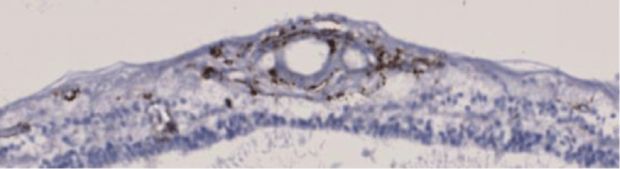
Between the ILM and the GCL inner boundary, the RNFL is constituted by RGC axons but also contains other cellular elements, including the inner retinal vascular branches of the central retinal artery and vein. The other constituents of the “RNFL” could include displaced amacrine cells, and Müller cell processes in the healthy state. (In fact, recent data suggests that up to half the cells of the GCL in the mouse may be amacrine cells rather than RGCs) [14]. In disease, infiltrating cells trapped anywhere between the ILM and GCL as well fibrosis/gliosis could also contribute volume to the RNFL[15]. In the pathology series 15% of patients had HLA-DR + cells found in the perivascular cuff (which would presumably be detected as RNFL) [Figure 14.4] [1], Outside of the peripapillary RNFL this would contaminate estimations of nerve fiber layer thickness of volume. However, this would only have limited impact on routine peripapillary RNFL thickness assessments because most perivascular sheathing has been identified to occur in the mid-periphery and not proximate to the optic disc [9, 11].
Immune cells in perivascular space. HLA-DR staining showing MHC class II positive immune cells adjacent to blood vessels in the nerve fiber layer of a subject with MS. Given the morphology, many of these cells appear to be lymphocytes.
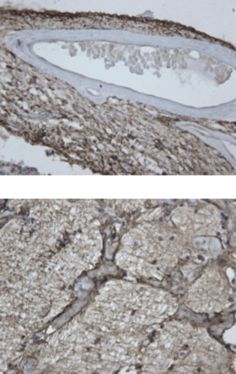
On the other hand, fibrosis especially at the optic nerve head was another frequently observed pathological phenomena seen in the pathology series [Figure 14.5]. This fibrosis was at times severe enough that appeared to lead to deformation of the normal architecture of the optic nerve head and could conceivably impact the shape of the cup and the volume of the RNFL [1, 4]. Additional, gliotic changes were detected using GFAP staining for astrocytes that showed reactive astrocytes next to vessels exhibiting blood retinal barrier disruption and located within the layer that constitutes RNFL [1].

Gliotic scar in the retina and optic nerve. Perivenular gliosis near optic disc (Panel A) and within optic nerve (Panel B) in subject with SPMS (GFAP stain)
Immune cells were not restricted to the RNFL in the study and HLA-DR positive cells with an activated appearance were found in deeper layers of the retina including the inner nuclear layer and inner plexiform layer [1]. Under normal circumstances microglial cells of the retina are tiled through the IPL and OPL surrounding the INL; however, in the state of activation they have the potential to migrate to sites of injury or inflammation [16].
One of the striking findings of the larger systematic evaluation of retinal samples from MS subjects was the identification of extensive injury involving the inner nuclear layer of the retina [1]. The population of neurons found in this layer (horizontal and bipolar cells) are not myelinated and, therefore, their loss suggests that there is either direct inflammatory mediated injury in the retina or that secondary trans-synaptic processes underlie cell loss in cells connected to a disrupted pathway.
In terms of the specificity of the findings this study had the added benefit that the evaluating pathologist was blinded to the underlying diagnosis (given embedded pathological controls). This served to reduce potential bias on the part of the evaluating pathologist in determining the frequency of the pathological features observed.
Neuronal ceroid lipofuscinosis
The neuronal ceroid lipofuscinoses are a set of inherited neurodegenerative diseases that exhibit significant progressive injury to the brain and retina. Considered together, they constitute the most common pediatric neurodegenerative disorders in humans with a cumulative prevalence of 1 per 10,000. Clinically, the diseases are characterized by developmental regression, visual disturbances, stimulus-induced myoclonus, seizures, and early death. The diseases typically manifest in the first decade of life – and the most common forms present with visual dysfunction – although later onset forms have been identified [17]. On ophthalmologic examination, most patients with NCL demonstrate significant optic atrophy and at least 70% have significant visual impairment at the time of death. Visual findings are particularly prominent in patients with infantile (CLN1) and juvenile (CLN3) onset NCL, where the injury is early and frequently profound. By contrast some of the other NCL subtypes, such as late infantile NCL (also known as CLN2), manifest visual symptoms and presumably retinal injury later in the disease. Most of the other NCL subtypes have also been described to have significant retinal injury – but are less common and the spectrum of disease has been less well described [17].
On brain imaging, NCL patients exhibit extensive confluent subcortical white matter injury along with severe brain atrophy and thalamic T2 hypointensities. The pathological characteristic feature of all these diseases is the intracellular accumulation of lipid- and protein-derived pigments called ceroid. Ceroid and lipofuscin are typically distinguished in that lipofuscin is lipopigment that accumulates as part of normal senescence but ceroid is lipopigment that accumulates as a consequence of a disease state. Both lipopigments and ceroid have the tendency to be endogenously autofluorescent, meaning that light of an appropriate frequency will elicit the release of photons with lower energy (i.e., a longer wavelength) [18]. In many instances, these autofluorescent materials are sensitive to a wide spectrum of stimulating light and, given their varied molecular structure, also emit a broad spectrum of photons in response.
The diagnosis of NCL is established through the identification of Periodic Acid Schiff and Luxol fast blue positive autoflourescent material in the brain and retina [17]. In these tissues, as well as in the muscle, subcutaneous fat, and skin, electron microscopy exhibits curvilinear bodies, fingerprint bodies, and Granular osmophilic deposits. Today, molecular diagnosis is the gold standard for confirming disease and establishing disease subtype. Given tissue accessibility and low morbidity of biopsy specimens, skin samples are typically the tissue of choice for disease identification and subtype confirmation can now frequently be confirmed via genetic testing.
Unfortunately, by the time of autopsy the disease has reached its end stage so that damage to neuronal and retinal populations is widespread. By some accounts, there is predominant injury to smaller cortical cells early, and injury in the retina and damage proceeds from the photoreceptor layers outwards. Clinical studies demonstrate retinal atrophy and pigmentary retinopathy that precede the development of nerve fiber layer loss of disc atrophy, suggesting the spread of disease to the inner retina [19, 20]. Histopathology from retinas on autopsy in Batten’s disease (now known as NCL type 3) showed total loss of photoreceptors, including both rods and cones and extending into the outer plexiform layer. Pigmentary changes were less uniform and the degree of injury to the ganglion cell layer were also less consistent [20]. Electron microscopy also demonstrated curvilinear and fingerprint bodies in the ganglion cells of NCL subjects.
In almost all described histopathology cases of NCL type 3 (Batten’s disease) there is accumulation of autofluorescent pigment in both retinal ganglion cells and cells of the inner nuclear layer. This distinguishes NCL from other ophthalmologic diseases with lipofuscin such as age-related macular degeneration in which lipofuscin accumulation is predominantly seen in the retinal pigment epithelium at the outer edge of the retina. Furthermore, lipopigment is not always found in cells that are degenerating – suggesting that lipofuscin/ceroid accumulation is not a necessary prerequisite for cell death [17, 20]. It should be noted, however, that only one autopsy case of NCL has been evaluated in the age of modern immunohistochemistry and that additional analyses and evaluations would be appropriate especially given the variety of genetic mutations described to date – even within a single disease causing gene. Furthermore, given the timing of tissue acquisition neither the precise evolution of tissue injury nor the cell types that are most susceptible to tissue injury is clear. Indeed, in CLN5, for example, there is not reported to be the “spread” of injury from the outer retina to the inner retina as described for CLN3 [17]. Furthermore, the number of retinas that have been pathologically described in the literature is small and a full characterization of the nature and extent of retinal injury remains to be performed.
Alzheimer’s disease
Alzheimer’s disease (AD) is the most common dementing illness of the elderly and accounts for more than half of dementia cases with pathological confirmation at autopsy. AD is pathologically characterized by neuron loss in specific areas of the brain as well as the presence of intraneuronal inclusions of hyperphosphorylated tau in vulnerable cell populations (known as neurofibrillary tangles) and plaques of amyloid beta (neuritic plaques). These amyloid plaques constitute the other pathological hallmark of AD and are composed of sheets of beta-amyloid peptides (these peptides in turn are 36–43 amino acids in length and are principally described by the peptide length detected).
Patients with AD have been reported to manifest visual dysfunction [21, 22], although the extent of these visual deficits has been difficult to characterize because of patients concomitant alterations in memory and behavior. There have been consistent reports documenting that patients with AD demonstrate significant inner retinal atrophy, including descriptions of disc pallor and nerve fiber loss documented with fundus photos and OCT [23–27]. Although most of the OCT studies were done on older technology the general impression has been that thinning predominantly involves the arcuate bundles rather than the papillomacular bundle. However, these studies have generally lacked pathological confirmation of diagnosis (and predated the use of amyloid PET-based imaging) and the total numbers of patients studied has been small.
A seminal paper in the field compared ten AD subjects with ten age-matched controls and described moderate levels of axonal degeneration in the optic nerves of subjects with AD. The investigators reported relatively selective loss of larger diameter axons in AD subjects (although very small axons of 0.5–1 micron diameter appeared to potentially be lost as well). Reactive gliosis was described in both the retina and the optic nerve [28]. The authors described particular diminution of the thickness of the nerve fiber layer. However, they specifically describe an absence of both intraneuronal tau and amyloid beta (as assessed by Thioflavin S staining) [29–31]. Ultrastructural assessments in a related set of samples reported a vacuolated frothy appearance to RGC cytoplasm. Using additional stains the investigators determined that in one case 20% of RGCs appeared to be actively degenerating [29]. Although the bulk of the literature in this area has suggested that inner retinal atrophy is detectable in AD, at least one well-documented series was unable to demonstrate any loss of RGCs or axonal loss in the retina [32].
The presence of amyloid beta fibrils in the retinae of AD patients is controversial and unclear. Most investigators have been unable to find amyloid deposits in the retinae of patients with AD. Age-related accumulation of amyloid beta has been described in the normal human and mouse retina [33], but only one group has reported the capacity to detect human retinal amyloid deposition in AD subjects [34, 35]. Others have found no evidence for either amyloid plaques or hyperphosphorylated tau in the AD retina [36].
In animal models of AD (mice genetically manipulated to recapitulate one or some of the major genetic defects associated with nonsporadic AD) pathological descriptions have suggested that Abeta deposition can be detected in the inner retina around blood vessels, and that this may be associated with ganglion cell loss and thinning of the retinal nerve fiber [34, 37]. This observation requires further replication and analysis.
Friedreich’s ataxia
Friedreich’s ataxia (FA) is an autosomal recessive neurodegenerative disease caused by excessive GAA repeats in intron 1 of the frataxin gene on chromosome 9. This leads to increased susceptibility to oxidative stress and mitochondrial dysfunction induced by aberrant iron influx. The disease is characterized by progressive leg weakness with ataxia and gait disturbance as well as loss of deep tendon reflexes and dysarthria. Up to 30% of patients with FA have been noted to manifest significant optic neuropathy [38, 39]. In addition, much less frequently patients exhibit progressive pigmentary retinopathy that appears similar to standard retinitis pigmentosa, and a different subset of patients has been reported to have more fuliminant and dramatic visual decline [38]. Although nearly all patients have evidence of injury to the anterior visual pathway, including thinning of nerve fiber, very few have significant visual symptoms. To date, there are no reported cases of pathological evaluation of the retina in patients with FA [38, 39].
Parkinson’s disease and synucleinopathies
Idiopathic Parkinson’s disease (IPD) is the second most common neurodegenerative disease in the industrialized world and is pathologically manifested by injury to dopaminergic cells of the central nervous system – most characteristically in the substantia nigra pars compacta of the midbrain. However, injury to the dopaminergic system is widespread in the disease and includes areas of the brainstem and cerebral hemispheres.
Induction of Parkinsonism in mice with the administration of 1-methyl-4-phenyl– 1,2,3,6-tetrahydropyridine (MPTP) produces a model of disease by selectively targeting dopaminergic function in the central nervous system. Use of MPTP causes a reversible but dose-dependent reduction in tyrosine hydroxylase in amacrine cells. In the absence of tyrosine hydroxylase, neurons cannot convert L-Tyrosine into L-DOPA (which is itself a necessary precursor for dopamine production) [40]. Decreases in retinal dopaminergic amacrine function might be predicted to result in failure of center surround antagonism in ganglion cells. Patients with IPD are known to have deficits detectable on flash electroretinograms and pattern electroretinograms – especially manifested in tests done with a high spatial frequency – as would be predicted by injury to amacrine cells. However, TH immunostaining in human subjects with IPD has been reported to be normal. Furthermore, intracellular aggregations of alpha-synuclein have been reported to be absent in the retina [41].
Despite interest in the field, and the recognition of visual functional deficits believed to be retinal in origin in subjects with IPD, there are no published descriptions of retinal histopathology in patients with IPD [36, 41]. A single pathological series including three patients has been the subject of two papers in patients with another major synucleinopathy – dementia with Lewy bodies. This analysis reported the identification of pale intracellular inclusions in the outer plexiform layer and cytoskeletal disorganization with enlarged rod inner segments. These inclusions were not synuclein, but their molecular structure was not identified [42].
Huntington’s disease
Huntington’s disease (HD) is a relentlessly progressive monogenic autosomal dominant hereditary neurodegenerative disease caused by expanded trinucleotide repeats in the gene encoding the protein huntingtin (Htt) on chromosome 4. The expanded CAG repeat gives rise to an excessively long polyglutamine region in mutant Htt. Disease onset is most typically in middle age and characterized by defects in motor function and psychiatric disturbances followed by the ultimate development of dementia and death. Principal injury occurs to the indirect pathway of the striatum including the caudate nucleus. A study of retinal function in living HD patients showed higher thresholds to blue light following adaptation to yellow light, suggesting deficient neural processing in the retina compared to both normal controls as well as with patients with Tourette’s syndrome and schizophrenia [43].
Murine and drosophila models of HD exhibit clear evidence of retinal degeneration as part of the disease phenotype. The R6/1 mouse model expresses exon 1 of the HD gene with 115 CAG repeats driven by the human HD promoter. Similarly, the R6/2 model expresses the same exon but with 150 repeats (and thereby has accelerated and more severe disease). The R6/1 model shows cone dysfunction and injury starting around the same age as other manifestations of disease in the brain. Ultimately, these mice showed complete loss of cone opsin and transducin. The more severe R6/2 model demonstrates significant injury on light and electron microscopy affecting the entire retina, especially concentrated in the center [44]. The drosophila model of HD also has evidence of photoreceptor neuron degeneration [45]. Only a single subject with HD appears to have ever undergone detailed ophthalmic pathological analysis. The investigators failed to find any clear retinal injury, but examination of the photoreceptor layer, which is affected in the drosophila model, was limited by technical difficulties because it peeled off the retina during processing [45].
Related posts:
 Optical coherence tomography and low-contrast acuity
Introduction to optical coherence tomography in neurological diseases
Future technological advances in optical coherence tomography
Optical coherence tomography in acute optic neuritis
Optical coherence tomography and visual outcomes in acute optic neuritis
Optical coherence tomography in a multi-center setting: quality control issues
Optical coherence tomography and low-contrast acuity
Introduction to optical coherence tomography in neurological diseases
Future technological advances in optical coherence tomography
Optical coherence tomography in acute optic neuritis
Optical coherence tomography and visual outcomes in acute optic neuritis
Optical coherence tomography in a multi-center setting: quality control issues
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


