RNFL thinning in NMO vs. MS.
Three examples of OCT findings from an MS patient’s eye with previous ON (left), a neuromyelitis optica spectrum disorder (NMOSD) patient’s eye with previous ON without microcystic macular edema (MME) (center), and an NMOSD patient’s eye with previous ON and MME (right). The first row shows peripapillary RNFL thickness data for average RNFL (G) and sectors. The second and third rows show thickness maps of the retinal ganglion cell layer (GCL) and inner nuclear layer (INL) (adapted from [15]).
In 2012, microcystic macular edema (MME) was reported in both MS and NMO in association with reduced visual acuity and retinal nerve fiber layer thinning [12, 15–17]. MME presents as a microcystic pattern with lower reflectance (Figure 11.2) and is mainly located in the inner nuclear layer (INL) in small, discrete patches [16]. Both the pathogenesis and association with various nosologic entities have yet to be clarified. One possibility is that MME is a symptom of chronic inflammation in the INL, because this layer was shown to be a prominent site of inflammation and microglia activation in a recent autopsy study [18]. A further argument for an inflammatory pathogenesis is provided by Saidha et al., who showed that the INL thickening, which usually accompanies MME, may be indicative of a more severe disease progression in MS, as indicated by a higher number of inflammatory lesions on brain MRI, accelerated EDSS progression, and a higher frequency of relapses [19]. A study by our group further revealed that MME is not specific to MS or NMO, but is strongly linked to a history of ON: 95% of eyes with MME (in total 22 eyes) from a cohort of 255 patients with MS or clinically isolated syndrome (CIS), 20 NMO spectrum disorder patients, and 9 patients with chronic inflammatory optic neuropathy had previously experienced ON. In all three groups, patients with unilateral ON and without visible MME showed an increased INL thickness of the ON-affected eyes compared to the contralateral eyes, suggesting that INL thickening and MME may both be symptoms of underlying retinal inflammation [20]. Additionally, Gelfand et al. only detected MME in eyes with a prior history of ON in a cohort of 25 NMO patients [17]. Both studies showed that MME may be more frequent in NMO (approx. 20%) as compared to MS (approx. 5%); however, this finding has to be interpreted with caution given the low number of the subjects characterized in the two NMO cohorts. In contradistinction to the premise that MME represents a cardinal feature of retinal edema and inflammation restricted to a few diagnostic entities, recent case reports on a patient with optic nerve glioma and a patient with Leber’s hereditary optic neuropathy (LHON), both of whom exhibited MME, strongly contested the idea that MME has an inflammatory pathogenesis, and instead suggested that MME may be a consequence of any optic nerve pathology, irrespective of their pathobiological basis. [21–23]
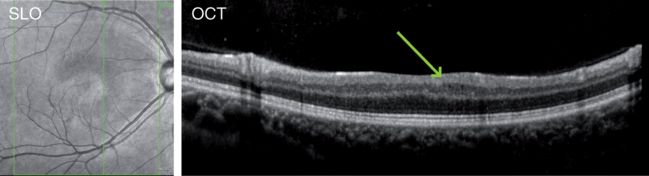
Microcystic macular edema (MME) in NMO
Examples of images from one NMO patient’s eye showing MME. In the scanning laser ophthalmoscopy (SLO) image on the left, MME is visible as the darker crescent around the fovea. In the OCT B-scan on the right, MME can be identified by the small hypointense lesions exclusively in the INL (green arrow).
The literature to date is inconclusive regarding the detection of retinal damage by OCT in NMO non-ON eyes. In some studies, NMO non-ON eyes exhibited no apparent retinal damage, as all OCT measurements closely approximated those derived from healthy control subjects [7, 8, 15]. This supports the notion that retinal damage in NMO is linked to clinically manifest ON attacks, and does not occur progressively or as a consequence of subclinical optic neuropathy, as may be the case in many MS patients [15]. Accordingly, NMO has rarely been found to exhibit features that would confirm the presence of a secondary progressive course [24]. However, Sotirchos et al. and Syc et al. have reported both GCL and IPL thinning in NMO non-ON eyes, a highly conspicuous and controversial finding that must be confirmed or refuted in future studies [11,12].
In summary, OCT data on morphological retinal damage in NMO-ON eyes and analysis of the correlation between this damage and poor visual function are abundant and largely consistent. In contrast, the pathophysiology of retinal damage in a disease characterized by serum antibodies against the CNS water channel AQP4 has not been fully elucidated. Mechanistic studies have shown that AQP4-antibodies not only serve as a biomarker for an NMO diagnosis, but also are pathogenically relevant in terms of binding to astrocytic membranes, the putative scaffolding upon which targeted damage to the aquaporin water channel can materialize [25, 26]. Could an inflammatory attack targeting the AQP4-expressing myelinated retrolaminar optic nerve conceivably cause retinal atrophy via a dying-back mechanism. A further and perhaps more intriguing scientific question arises from evidence that the retina also contains a plethora of astrocytes that express AQP4 at high levels (e.g., Müller cells are but one of a number of astrocytic subtypes capable of cell-surface expression of this key target antigen within the retina). Taken together, the evidence would suggest that the retinal Müller cell represents a bona fide second ocular target of pathogenic AQP4 antibodies [27]. Confirmation of this potentially seminal observation represents a formidable challenge, particularly given that adequate animal models for human NMO have yet to be developed.
In the clinical setting, the use of OCT in the diagnosis and treatment of NMO is proliferating throughout the neurological and ophthalmological communities, with the powerful advent of performing a simple, rapid, convenient, reproducible, and noninvasive assessment of an elegantly eloquent CNS compartment, with compelling ramifications for longitudinal ascertainment of retinal damage, and its corresponding relationship to clinical disability, along with emerging utility as a tool for differentiating those pathophysiologic mechanisms that help distinguish NMO from MS and other conditions. Not surprisingly, the features that characterize high precision, high-speed, high-definition spectral-domain OCT have ushered in a new and exciting era, one where this powerful technique may have utility in helping to advance treating neuroinflammatory and neurodegenerative disorders by being endowed with the capability to detect and monitor preventative, protective, and even restorative properties of novel neurotherapeutic agents.
Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative condition that typically presents with motor symptoms such as akinesia, tremor, and rigidity, and olfactory dysfunction. However, PD has also been widely reported to include visual symptoms like decreased contrast sensitivity and color discrimination, visuospatial deficits, visual hallucinations [28–30], and some electrophysiological studies of visual evoked potentials (VEPs) and pattern electroretinograms suggest pathological signatures within the retina and/or the anterior visual pathways [30–32]. While PD has long been associated with a neuropathological hallmark, the accelerated loss of dopaminergic (DA) neurons within the substantia nigra, dopaminergic neurons (amacrine cells) have also been detected in the INL and IPL of the human retina, thereby suggesting that the pathobiological basis for PD involves, at least in part, the specific targeting of DA neurons by disease-specific injury cascades [33].
Retinal DA deficiency may contribute to visual impairment in PD, as DA is involved in visual processing by modulating the organization of receptive fields of the retinal ganglion cell neurons, whose axons ultimately coalesce into the optic nerve [34, 35]. A postmortem study showed decreased DA in retinas from PD patients who had not been treated with levodopa [36], whereas an in vivo study involving PD patients reported improvement of spatial contrast sensitivity after treatment with levodopa [37].
Several OCT studies have investigated retinal atrophy in patients with PD with diverging results, probably as a result of differences between the cohorts and the OCT devices used. The validity of most OCT studies in PD is hampered by small cohort size and the insufficient or ambiguous consideration of confounding factors prevalent among PD patients, including glaucoma and diabetic retinopathy, to name a few. Altintas et al. found a significant reduction in average RNFL thickness and TMV in 17 PD patients vs. healthy controls, and an inverse correlation between foveal retinal thickness and the Unified Parkinson Disease Rating Scale (UPDRS), a clinical scale to assess neurological impairment in PD [38]. In another investigation, Inzelberg et al. reported a reduction of RNFL thickness in inferior and temporal quadrants in 10 PD patients [39], while a another study found a decreased foveal thickness in 9 PD patients [40]. Hajee et al. demonstrated significant thinning of the inner retinal layers in 24 PD patients [41], and Aaker et al. found differences in macular thickness (thinning and thickening), but no differences in RNFL thickness in 9 PD patients vs. 16 controls [42]. Shrier et al. found increased intraocular differences in macular volume between 23 PD patients and 18 controls [43]. In contrast, Archibald et al. did not find any alteration in RNFL thickness or TMV in 37 PD patients [44]. Likewise, application of modern SD OCT technology in another study showed no differences in RNFL and TMV between 40 PD patients and 25 healthy controls [45]. Interestingly, manual intra-retinal segmentation revealed a thicker INL in PD vs. controls in this study. No correlation between OCT measures and visual acuity, disease duration, or disease severity according to the UPDRS was observed. In a recent study using SD OCT, Garcia-Martin et al. investigated 75 PD patients in comparison with 75 matched healthy controls and found subtle RNFL reduction using three different RNFL scan protocols [46].
Our group recently completed a spectral-domain OCT study on 97 patients with idiopathic PD and 32 healthy controls [47]. Intra-retinal segmentation revealed significant thinning of the combined outer nuclear (ONL) and photoreceptor layer (PRL) in PD vs. controls (118.6 vs. 123.5 µm, p = 0.001). Neither RNFL thickness nor TMV or any of the other layers differed from those of controls. We investigated whether retinal damage is associated with impaired color vision, as assessed by the Farnsworth-Munsell color discrimination test (FMT), as color perception has been reported to be altered in PD, a salient feature that may be associated with disease progression [48]. We identified no correlation between performance in the FMT, and any of the OCT measures. Furthermore, no association was found between OCT measures and clinical parameters such as UPDRS or disease duration. Despite the heterogeneity of previous OCT results, our study, in which we had applied modern SD OCT technology with intra-retinal segmentation, as previously used in numerous studies [49], clearly showed that retinal DA deficiency may cause structural retinal damage beyond the RNFL that is detectable by OCT. The cause for this observation remains largely unexplained, and we have already formulated strategic plans to fully characterize this pathological substrate. For example, the PRL contains rods and cones that receive input from dopaminergic interplexiform and amacrine cells via the inner and outer plexiform layers [50, 51]. Thus, we may assume a process of transsynaptic degeneration of ONL neurons as a consequence of altered synaptic input from dopaminergic neurons. The fact that atrophy of the ONL/PRL was not associated with deficits in color discrimination may be – at least in part – explained by the observation that FMT is prone to various confounding elements such as motor and cognitive impairment as well as dopaminergic treatment.
In summary, OCT data in PD are highly heterogeneous, and the value of OCT in the clinical assessment of patients with PD remains to be established. A complicating factor is that many PD patients have difficulties complying with the experimental requirements of OCT examination (thereby leading to a clear acquisition bias) due to head or neck tremor, axial rigidity, or cognitive impairment. Recognizing these important and test-limiting factors may represent at least part of the bases for the discrepant outcomes across previous studies. Ultimately, OCT image quality from PD patients has been demonstrated to be of lower than the image characteristics derived from healthy controls. Image quality cannot be ignored, especially given that OCT generated data can easily be skewed in the setting of poor-quality images [52].
Alzheimer’s disease
Alzheimer’s disease (AD), which predominantly affects episodic memory, is the most common neurodegenerative cause of dementia in the elderly. The pathophysiological cause is unclear, but the deposition of Aβ plaques in the brain has been suggested as having a major role and is linked – along with other biomarkers – to clinical disease severity [53]. Recently, the presence of Aβ plaques in the retina was reported in a murine Alzheimer’s disease model, and confirmed in postmortem tissue from Alzheimer’s patients. Importantly, retinal Aβ plaques correlated with brain pathology and clinical severity, and were detectable in suspected AD patients [54]. These recent observations might lead to AD-specific retinal imaging, once labeling of the pathognomonic Aβ plaque becomes feasible in human subjects. The authors of this study (ref 54) have provided the first in vivo data in mice using systemically administered curcumin for the specific labeling of Aβ plaques.
Currently, researchers and clinicians seeking to focus on the retina of AD patients face two challenges. First, although visual symptoms in AD patients are widely reported, they could easily be the result of impairment to higher cognitive functions necessary for visual processing [55, 56]. Second, the high coincidence of other pathologies commonly associated with the aging retina problematizes analysis. Age-related macular dystrophy (AMD), vascular changes, wide-angle glaucoma, cataract, and diabetic retinopathy are common comorbidities that partially account for the reported visual dysfunction in AD (see also below). Consequently, distinguishing between retinal changes that are potentially AD-related and those caused by comorbidities is difficult.
Studies investigating retinal changes in AD have yielded conflicting results: A reduction of retinal ganglion cells (RGC) in comparison to healthy controls was reported in an early postmortem study investigating 10 AD patients [57], as well as in a further study comprised of 9 AD patients with matched healthy subjects [58]. RGC reduction was confirmed by investigating postmortem optic nerve tissue from AD patients, what showed prominent loss of M-cells, the largest class of RGCs contributing large calibre fibers to the optic nerve [59]. Other histopathological studies reported no differences in the layout in the AD patient’s retina [60–62]. The latter is supported by several studies using scanning laser polarimetry, retina tomography, and pattern electroretinography [62–66].
Several studies have applied OCT to investigate retinal changes in AD patients. Iseri and colleagues investigated 14 AD patients vs. 15 matched healthy subjects. They reported a reduction of RNFL (AD, average RNFL 87±24 µm (mean ± standard deviation) vs. controls, average RNFL 113 ±7 µm) and total macula volume (AD, TMV 6.8 ±0.4 mm3 vs. controls, TMV 7.1 ±0.2 mm3). Reduction in TMV correlated with clinical severity expressed by mini-mental state examination [67]. Paquet et al., who in addition to AD patients also investigated cohorts with mild cognitive impairment (a precursor symptom of AD), reported a similar finding in 49 cognitively impaired patients. Here, RNFL reduction was more prominent in the more severely affected patients [68]. Lu et al. reported RNFL thinning in the superior and inferior quadrants in 22 patients compared to age-matched healthy controls [69]. Berisha and colleagues studied nine early stage AD patients vs. matched healthy subjects and found that the AD patients showed significant RNFL reduction in the superior quadrant (AD, superior RNFL 92 ±22 µm compared to the controls’ superior 114 ±1 µm) [70]. In one of the largest AD cohorts investigated to date, which included 40 patients and 40 well-matched healthy subjects, Kirbas and colleagues reported significant RNFL reductions (AD, average RNFL 65 ±6 µm vs. average RNFL 75±4 µm). Again, RNFL thinning was predominantly located in the superior quadrant [71].
In summary, these OCT studies suggest significant RNFL reduction is a common feature of AD. As in PD, owing to comorbid physical and cognitive symptoms, performing OCT examinations reliably on AD patients can represent a formidable challenge, one that markedly limits obtaining data of acceptable quality, whether used in clinical practice or as outcome measures in clinical research. The recognized high standard deviations of the data collected from small cohorts of patients afflicted by neurodegenerative disease may reflect the broad pathological heterogeneity across patients carrying the same diagnosis. Further, the age matching in most of the referenced studies lacked stringency, adding age as a potentially important confounder.
Of note in this regard is another important finding in AD: the high co-occurrence with open-angle glaucoma (OAG). In comparison to healthy subjects, AD patients show a higher occurrence of OAG [72], and subjects with OAG show a higher risk to develop AD in later life [73]. The link between OAG and AD is currently unclear and needs to be investigated further in the future. Conspicuously, the preponderance of RNFL loss in the superior quadrant in most of the studies interrogated may also represent a potential confounding effect, namely, undiagnosed glaucoma, which also tends to affect the superior quadrant [74].
Other neurologic diseases
Susac syndrome
Susac syndrome presents as a combination of encephalopathy, hearing loss, and visual deficits due to acute branch retinal artery occlusions [75]. The etiology of this rare disease and its exact prevalence are unknown [76]. Differentiating it from MS can be challenging since the corresponding clinical presentation and diagnostic findings may significantly overlap. However, in OCT, eyes from patients with Susac syndrome show sectorial damage that can be distinguished from MS patients [77]. During an acute event of Susac syndrome, the affected areas show profound swelling, which later leads to almost complete loss of all inner retinal layers supplied by the occluded branch artery (Figure 11.3). This presentation during the acute phase of visual symptoms clearly differentiates Susac syndrome from optic neuritis resulting from MS or NMO, where acute and chronic damage is much more broadly distributed and certainly not a consequence of a single vessel distribution of localized tissue damage.
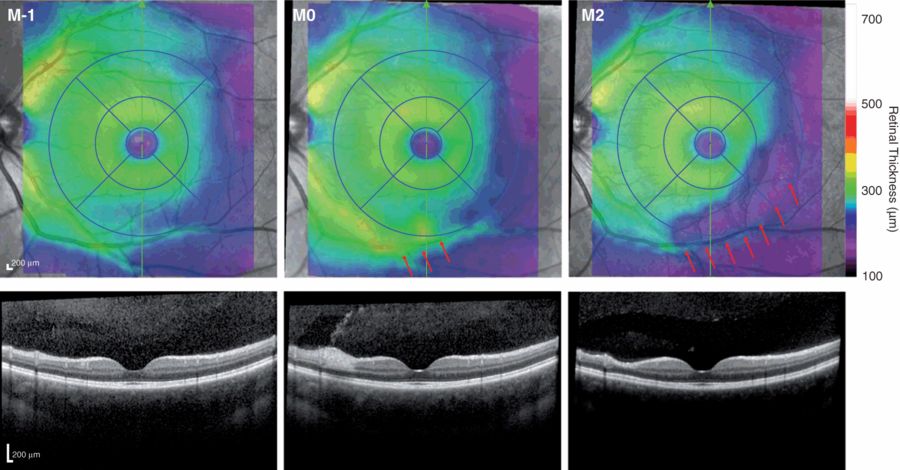
Formation of a Susac syndrome retinal lesion.
Examples of findings from a Susac syndrome patient’s eye. One month prior to the acute lesion (M-1 on the left), the retina appears mostly normal in the nasal-inferior area around the macula. During the acute event (M0), massive swelling can be observed around the retinal branch vessel directly inferior to the macula, indicated by the red arrows. After two months (M2 on the right), the swelling has decreased and the inner retinal layers at the site of prior inflammation and in all of the area supplied by this vessel are almost completely degenerated, indicated by the red arrows.
In differential diagnosis – for example, when acute visual symptoms present in combination with cognitive changes or hearing loss – OCT should always be performed to exclude possible Susac syndrome. A suspected diagnosis of Susac syndrome can then be substantiated using fluorescence angiography (FAG).
Intracranial hypertension
Elevated intracranial pressure can present as papilledema. This edema of the optic nerve head (ONH), long familiar to neurology and ophthalmology clinicians, can also be measured using OCT (Figure 11.4) [78]. In idiopathic intracranial hypertension (IIH), our group could show that ONH volume measurements can reliably quantify the papilledema and discriminate between healthy subjects, IIH patients undergoing clinically effective therapy but who nevertheless exhibited increased ONH volume, and untreated patients, who showed the largest ONH volume [79]. This parameter (i.e., ONH volume) correlated highly with intracranial pressure (ICP), suggesting that OCT is also of benefit in the diagnosis and monitoring of patients affected by other diseases with elevated ICP. Another study using the Frisén scale, which quantifies edema using fundus photography, also showed good correlation between an alternative ONH quantification approach and Frisén scale derived papilledema grades [80]. Currently, further studies are needed on the temporal dynamics in conjunction with inter- and intra-individual changes in ONH volume, and visual function loss and ICP. Such studies will determine whether OCT has satisfactory utility as a surrogate biomarker in the assessment of ICP.
| SCA-3 | SCA-6 | SCA-7 | MSA-C | ARSACS | ALS | Wilson’s Disease |
|---|---|---|---|---|---|---|
| Pula et al. [90] | Pula et al. [90] | Manrique et al. [91] | Pula et al. [90] | Vingolo et al. [95] | Roth et al. [101] | Albrecht et al. |
| 2011 | 2011 | 2009 | 2011 | 2011 | 2013 | 2012 |
| Spectralis SD-OCT | Spectralis SD-OCT | Stratus TD-OCT | Spectralis SD-OCT | not reported | Cirrus HD-OCT | Spectralis SD-OCT |
| 5 (n=29, 58 y, 18/11)* | 6 (n=29, 58 y, 18/11)* | 7 (5 from one family) | 5 (n=29, 58 y, 18/11)* | 5 (34±8 y, 5/3) | 76 (56±11 y, 26/50) | 42 (40±14 y, 24/18) |
| 27 HC (56 y, 18/9)* | 27 HC (56 y, 18/9)* | none | 27 HC (56 y, 18/9)* | 5 HC (62±8 y, 4/1) | 54 HC (56±11 y, 26/28) | 76 HC (43±13 y, 35/29) |
| Significant RNFL reduction (85±9 µm) vs. HC (98±9 µm). | Non-significant RNFL reduction (95±5 µm) vs. HC (98±9 µm). Significant reduction of macular thickness (310±5 µm vs. HC (339±17 µm). | Case-wise discussion. RNFL reduction with focus on the superior and in more severe cases also on nasal and inferior quadrants. | Non-significant RNFL changes (100±11 µm) vs. HC (98±9 µm). Significant reduction of macular thickness (314±12 µm) vs. HC (339±17 µm). | Case-wise discussion. RNFL thickening with myelinated appearance. | No changes in RNFL (94±9 µm) vs. HC (93±10). No differences in GCL, INL/OPL and ONL/PRL. Only 15 definite ALS according to El Escorial criteria. | Significant RNFL reduction (95±9 µm) vs. HC (97±10). Reduction in GCL and INL. |
| Significant RNFL reduction. | Significant macular thickness reduction but no changes in RNFL. | Mild to severe RNFL loss focussed on superior, nasal and inferior quadrants. | No changes. | RNFL thickening with myelinated appearance. | No changes. | RNFL, RGC and INL thinning. |
* Pula et al [90]. only reported cohort statistics for the full cohort, not for disease subtypes
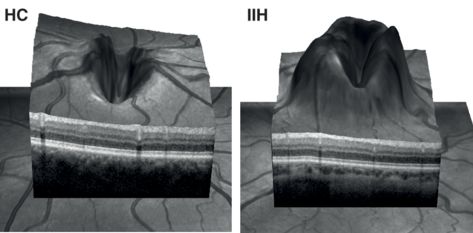
Papilledema in idiopathic intracranial hypertension.
Comparison of a 3-D optic nerve head scan from a healthy subject (HC on the left) and a patient with idiopathic intracranial hypertension (IIH) with increased ICP (adapted from [79]).
Hereditary and other rare neurodegenerative diseases
OCT has been applied in several other hereditary or rare neurodegenerative diseases. With a few exceptions, most published OCT studies show varying levels of RNFL reduction in the peripapillary ring scan that is often accentuated in the temporal quadrant. Fibers of the temporal quadrant RNFL comprise primarily parvo-cellular axons from the papillo-macular bundle. These axons consist of small and thinly myelinated axons with rapid firing rates that connect macular ganglion cells via the lateral geniculate nucleaus with the visual cortex. As such, they are highly relevant to high-resolution visual acuity and high spatial frequency of contrast sensitivity and color vision. Degeneration of these macular fibers has a much greater effect on visual quality of life than defects in the peripheral visual areas. Unfortunately, precisely these fibers are affected by neurodegeneration in many mitochondriopathies and other optic neuropathies, such as Leber’s hereditary optic neuropathy (LHON) and OPA1-related dominant optic nerve atrophy (DOA) [81]. One reason for this may be their small volume and fast firing, which might make them more susceptible to energy depletion by defective cellular metabolism and, thus, neurodegeneration [82]. Consequently, OCT assessment of RNFL focuses on the peripapillary ring rather than the macula. An overview of current OCT studies is given in Table 11.1, and a summary of key findings is provided in the following paragraphs.
| Susac | FRDA | SCA-1 | SCA-2 | |||
|---|---|---|---|---|---|---|
| Study | Brandt et al. [77] | Fortuna et al. [84] | Noval et al. [86] | Stricker et al. [89] | Pula et al. [90] | Pula et al. [90] |
| Year | 2011 | 2009 | 2012 | 2010 | 2011 | 2011 |
| Device | Stratus TD-OCT | Stratus TD-OCT | Stratus TD-OCT | Spectralis SD-OCT | Spectralis SD-OCT | Spectralis SD-OCT |
| Cohort (age, F/M) | 9 (33±11 y, 6/3) | 26 (32±8 y, 8/18) | 23 (25±7 y) | 9 (52±9 y, 4/5) | 7 (n=29, 58 y, 18/11)* | 7 (n=29, 58 y, 18/11)* |
| Matched cohorts (age F)/M | matched HC and MS | 48 HC (33±8 y, 20/28) | none | 9 HC (51±9 y, 4/5) | 27 HC (56 y, 18/9)* | 27 HC (56 y, 18/9)* |
| Main results | Significant RNFL reduction (81±18 µm) vs. HC (107±9 µm). in comparison to MS sectorial damage both in RNFL and TMV. | Significant RNFL reduction (76±12 µm) vs. HC (100±9 µm). Description of 3 distinct loss types correlating visual dysfunction. | RNFL reduction in comparison to normative data and with good correlation to visual function tests. | Significant reduction of average RNFL (84±13 µm) and temporal RNFL (62±8 µm) vs. HC average RNFL (97±8 µm) and temporal RNFL (74±9 µm). | Non-significant RNFL reduction (93±8 µm) vs. HC (98±9 µm). | Significant RNFL reduction (84±12 µm) vs. HC (98±9 µm). |
| Summary | Sectorial damage separates Susac retinal damage from ON related retinal damage. | RNFL reduction in 3 distinct types correlating with visual function. | RNFL reduction. | RNFL reduction with temporal quadrant focus. | No changes in RNFL. | Significant RNFL reduction. |
Friedreich’s ataxia
One of the first hereditary ataxias investigated in detail using OCT was Friedreich’s ataxia (FRDA) [83]. Visual symptoms and optic neuropathy are regularly found in FRDA [84] and mitrochondriopathy is likely to directly contribute to the disease pathophysiology [85]. In OCT, FRDA patients present with widespread RNFL reduction. Notably, visually asymptomatic patients also show optic neuropathy, the level of which correlates to general clinical severity and progression [84, 86, 87]. Fortuna et al. proposed three different types of RNFL degeneration in FRDA: Type 1 with diffuse and severe reduction of RNFL thickness in all quadrants, Type 2 with diffuse RNFL reduction that is more marked in the superior quadrant, and Type 3 with diffuse but mild reduction of RNFL thickness in all quadrants [84]. However, the clinical and pathophysiological significance of the individual categories remains to be elucidated.
Spinocerebellar ataxias
Spinocerebellar ataxias (SCAs) comprise a group of over 30 genetically distinct hereditary ataxias with similar clinical features. They are mostly numbered according to the date of their first description. Differential diagnosis of these disorders can be challenging because patients often present with only a subset of the symptoms known for SCA diseases as a whole [88]. OCT can aid in differential diagnosis by identifying patients with optic nerve atrophy especially without apparent visual symptoms. However, in many diseases OCT changes might be statistically significant but hardly relevant on an individual level.
Retinal changes identified by OCT have been reported in SCA patients with and without visual symptoms. SCA-1 presents with a mild and temporally focused RNFL reduction, and exhibits features similar to other mitochondria-related optic neuropathies [89]. SCA-2, SCA-3 and SCA-6 patients also show RNFL or macular thickness reductions [90]. The focal distribution of changes in many SCAs is currently unclear, as is a potential correlation with clinical scores, because most studies have involved only a few patients, especially in the rare SCAs, which limits their usefulness for detailed review.
Of the forms of SCA, SCA-7 is unique in having a high prevalence of retinal photoreceptor abnormalities that appear to be directly related to disease pathology. Consequently, SCA-7 patients regularly present with visual symptoms. However, current OCT technology can only detect these photoreceptor anomalies with difficulty and primarily in advanced disease stages when RGCs and retinal axons in the RNFL undergo secondary neurodegeneration [91]. In contrast to the temporal quadrant RNFL effect usually found in mitochondria-related disorders and other SCAs [92, 93], in SCA-7 patients’ eyes, the superior, inferior, and nasal quadrants are preferentially affected, whereas the temporal quadrant is mainly affected during late stages of disease [91].
Preliminary data on OCT findings further exist in SCA-14. In the largest cohort of SCA-14 patients investigated to date (n = 15) – a SCA that does not commonly present with visual symptoms – there were no SCA-14 specific retinal changes detectable on testing (unpublished data).
Autosomal recessive spastic ataxia of Charlevoix-Saguenay
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a rare disorder that was first identified in the Quebec region but can today be found throughout Northern America and Europe [94]. In OCT, patients with ARSACS present with increased RNFL thickness [95], which has been proposed as due to pathological myelination of retinal axons [96] or axonal hypertrophy (Figure 11.5) [97].
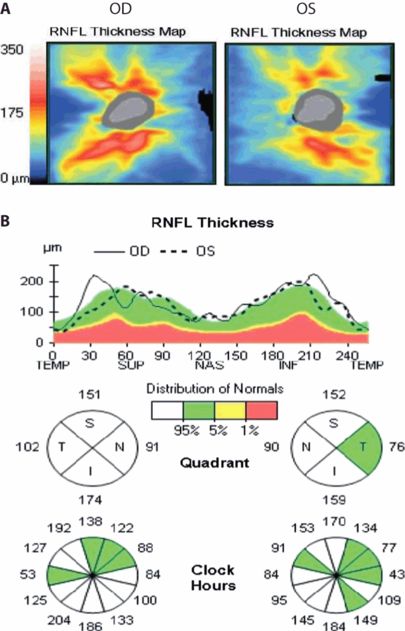
OCT findings in ARSACS.
Examples of an OCT report from an ARSACS patient. (A) RNFL thickness maps around the optic nerve head show RNFL thickening; (B) RNFL thickening can also be detected using the peripapillary RNFL ring scan. Comparison to RNFL distribution of normal controls shows elevated RNFL thickness in the inferior and superior quadrants (kindly provided by Dr. Elena Garcia-Martin, Zaragoza, Spain).
Hereditary spastic paraplegia
Hereditary spastic paraplegias (HSPs) are progressive neurodegenerative disorders with several distinct genetic phenotypes [98]. Common features are a dysfunction of axonal transport, predominantly leading to axonal length-dependent neurodegeneration of corticospinal tracts. Visual symptoms are rarely reported and mostly are limited to complex forms of the disease [99]. OCT shows RNFL reduction in temporal quadrants in clinically complex forms of the disease but not in clinically pure forms [100].
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting upper and lower motor neuron tracts. Although visual symptoms are uncommon, VEP abnormalities have been reported. However, there is currently no evidence that OCT can detect retinal involvement in amyotrophic lateral sclerosis (ALS) [101]. This requires confirmation in further studies, because genetically distinct subtypes of this heterogeneous disease, which has several different clinical phenotypes, might lend themselves to assessment by OCT.
Wilson’s disease
Wilson’s disease (WD) is a hereditary disorder of copper accumulation that leads to hepatic damage and neurodegeneration [102]. Although only few patients report visual symptoms, many patients show pathological findings in electroretinography and VEP [103]. In OCT, patients with WD show reduction in RNFL and RGC thickness [104].
Limitations
While OCT is likely helpful to further elucidate the natural or treated history of neurodegenerative disease, or to complete models of structure–function correlation for treatment, OCT measurements are not specific for any particular disorder.
First, RNFL changes with age. In healthy subjects, annual RNFL decrease is reported as between –0.16 µm/year [105] and –0.44 µm/year [106]. However, this decrease is not evenly distributed over age but is initially slow and accelerates after the age of 50 [105, 106]. A similar process takes place in the macula and affects mainly the RNFL, RGC, IPL, and INL but not, it appears, the outer plexiform and nuclear layers, including the photoreceptors [107]. Age-related accelerated loss of RNFL and macular thickness in older patients makes OCT assessment of optic nerve or retinal neurodegeneration in neurodegenerative diseases more suitable in younger patients. In clinical research, exact age matching of cohorts is, therefore, essential to ensure the validity of results. Minor differences in age can produce differences in results unrelated to the neuropathology being studied. Additionally, the coincidence of other eye diseases, particularly glaucoma (GD) but also diabetic retinopathy (DR) and AMD, increases with age. Above all, the risk of GD and here open-angle glaucoma rises considerably after the age of 50 [108]. While open-angle glaucoma prevalence among 50-year-olds is only 0–3%, in 80-year-olds it rises to 5–15%. Its incidence is also strongly ethnicity-dependent, with the disorder being more common among African, Asian, and Latin American groups than Caucasians [109]. Moreover, GD often also remains undiagnosed [110] because retinal changes due to GD are sometimes difficult to differentiate from reported findings in several neurodegenerative diseases. This makes GD an important confounder that should be considered in both research and clinical routine. Although some OCT studies discussed here went to considerable lengths to minimize potential confounder effects especially all of the studies investigating older cohorts should be interpreted with these limitations in mind.
Second, the majority of existing studies, especially in rarer neurodegenerative diseases, are cross-sectional studies, which often included only a small number of subjects. Only in very few diseases, such as NMO and SCA-1, have findings been reported and confirmed by several independent groups. In all cases, the cross-sectional nature of the studies makes validating the disease specificity of many findings difficult. Longitudinal studies are urgently required to better implement disease-specific and neurodegeneration-generic OCT findings into clinical routine.
Finally, OCT examinations are more demanding in severely clinically affected patients, who have difficulty complying with the needs of the experimental setting due to cognitive or motor impairment. In clinical routine, and especially in research, strictly adhering to the quality guidelines given in literature is vital to preventing artifacts and diagnostic errors [111]. Any significant variation in image quality should be investigated and reported [52].
Summary
OCT has potential benefit in the assessment of several neurodegenerative diseases. In routine clinical practice, OCT is highly recommended for both differential diagnosis and follow-up of patients with (suspected) MS and related neuroimmunologic diseases like NMO or Susac syndrome. Above all, OCT should always be performed as part of the differential diagnosis of patients presenting with symptoms indicating acute optic neuritis [112].
Furthermore, using OCT to detect optic nerve atrophy, which is characteristic of some, but not all, neurodegenerative diseases is of benefit in the differential diagnosis of other complex, often-related diseases. It provides an objective means of detecting visual system involvement, which is challenging to assess and often overlooked, particularly in patients with cognitive impairment. In our experience, OCT is a means of tracing visual symptoms in these patients to retinal or optic nerve abnormalities. Therefore, we suggest routinely performing OCT if at least one important differential diagnosis involving the retina is under discussion for a patient. Moreover, several rare diseases show distinct retinal pathologies that can easily be identified by means of OCT.
The current value of OCT in AD and PD is far more difficult to assess. Both diseases show clear retinal pathologies – in the case of AD the formation of possible retinal Aβ plaques and in the case of PD neurodegeneration of dopaminergic ganglion cells and potentially also photoreceptors. However, state-of-the-art OCT is currently unable to trace these changes, which are probably simply too subtle. Higher imaging resolution or functional OCT macular scan applications that specifically target the affected structures in these diseases might well solve this limitation in the future.
Currently, both diseases show a high co-occurrence with other diseases or changes of the aging retina, such as GD DR, AMD, and vascular changes. This makes applying OCT challenging, particularly when attempting to track disease-specific changes. More importantly, such confounders increase the quality requirements for OCT studies on AD and PD tremendously and hamper assessing the validity of published studies. Current data do suggest OCT is a valuable tool for studying AD and PD in research settings. However, clinician researchers should interpret possible findings cautiously, keeping the preceding issues in mind.
We expect OCT will prove a valuable follow-up marker in many of the diseases discussed previously. However, more research is required into its role as a disease monitoring tool before we can support its integration into everyday routine clinical practice.
Acknowledgments
We thank Ella Maria Kadas and Timm Oberwahrenbrock for assistance with preparing the figures. This work was supported by the German Research Council (DFG Exc 257 to FP) and the German Ministry for Education and Research (KKNMS Competence Network Multiple Sclerosis to FP).
References
Related posts:
 Optical coherence tomography and low-contrast acuity
Introduction to optical coherence tomography in neurological diseases
Future technological advances in optical coherence tomography
Optical coherence tomography in acute optic neuritis
Optical coherence tomography and visual outcomes in acute optic neuritis
Optical coherence tomography in a multi-center setting: quality control issues
Optical coherence tomography and low-contrast acuity
Introduction to optical coherence tomography in neurological diseases
Future technological advances in optical coherence tomography
Optical coherence tomography in acute optic neuritis
Optical coherence tomography and visual outcomes in acute optic neuritis
Optical coherence tomography in a multi-center setting: quality control issues
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


