In 1817, James Parkinson described the major clinical motor features of what today is recognized as the symptom complex known as parkinsonism, manifested by any combination of six cardinal features: tremor at rest, rigidity, bradykinesia-hypokinesia, flexed posture, loss of postural reflexes, and the freezing phenomenon. At least two of these features, with at least one being either tremor at rest or bradykinesia, must be present for a clinical diagnosis of parkinsonism. The many causes of parkinsonism (Table 83.1) are divided into five categories—primary, symptomatic/secondary, Parkinson-plus syndromes, various heredodegenerative diseases in which parkinsonism is a manifestation, and parkinsonism with neurotransmitter enzyme deficit. An example of parkinsonism with dopamine enzyme deficit and a benign clinical course is the condition known as dopa-responsive dystonia, covered in the chapter on dystonia (see Chapter 76). Primary parkinsonism is known as Parkinson disease (PD), which can be sporadic or familial; it is the most common type of parkinsonism encountered by neurologists and is the second most common neurodegenerative disease after Alzheimer disease (AD). In addition to the motor features of PD, the importance of nonmotor features, ranging from fatigue, sleep, and behavioral disturbances to autonomic and sensory symptoms is increasingly appreciated as contributors to the overall disability of patients with PD.
The core biochemical pathology in parkinsonism is decreased dopaminergic neurotransmission in the basal ganglia. In most of the diseases in Table 83.1, degeneration of the nigrostriatal dopamine system results in marked loss of striatal dopamine content. In some, degeneration of the striatum with loss of dopamine receptors is present and is probably responsible for the lack of therapeutic effect by dopaminergic agents in these disorders. Drug-induced parkinsonism is the result of blockade of dopamine receptors or depletion of dopamine storage. It is not known how hydrocephalus or abnormal calcium metabolism produces parkinsonism. Physiologically, the decreased dopaminergic activity in the striatum leads to abnormal activities of the subthalamic nucleus and the globus pallidus interna (GPi), which is the predominant efferent nucleus in the basal ganglia. Understanding the biochemical pathology led to dopamine replacement therapy; understanding the physiologic change in brain network circuitry led to surgical interventions, such as deep brain stimulation of GPi, the subthalamic nucleus and ventrointermediate nucleus of the thalamus. Lesioning of these nuclei (pallidotomy, thalamotomy) is an alternative surgical technique. In addition, nondopaminergic system deficit is associated with many of the nonmotor symptoms.
EPIDEMIOLOGY
PD makes up approximately 80% of cases of parkinsonism listed in Table 83.1. The incidence and prevalence of PD increase with age. The mean age at onset is about 60 years. Onset at younger than 20 years is known as juvenile parkinsonism. Many cases are due to mutations in the PRKN gene, an autosomal recessive disorder without Lewy bodies in the degenerating substantia nigra. Some heredodegenerative diseases such as Huntington disease and Wilson disease can present as juvenile parkinsonism. Onset of primary parkinsonism between 20 and 40 years is defined as young-onset PD; some investigators extend the age to 50 years to account for higher genetic contribution than later onset cases. PD is more common in men, with a male-to-female ratio of 3:2. The prevalence of PD is approximately 160 per 100,000, and the incidence is about 20 per 100,000 per year. The prevalence and incidence increase exponentially with age, and at age 70 years, the prevalence is approximately 550 per 100,000, and the incidence is 120 per 100,000 per year.
PATHOBIOLOGY
PATHOLOGY
The pathology of PD is distinctive. Degeneration of the neuromelanin-containing neurons in the brain stem occurs, especially dopamine-containing neurons in the ventral tier of the pars compacta in the substantia nigra and in the noradrenergiccontaining neurons in the locus ceruleus; many of the surviving neurons in these nuclei contain eosinophilic cytoplasmic proteinaceous inclusions known as Lewy bodies, the pathologic hallmark of the disease. The nigral dopaminergic neurons project to the neostriatum (nigrostriatal pathway). By the time symptoms appear, the substantia nigra already has lost about 60% of dopaminergic neurons and the dopamine content in the striatum is about 80% less than normal. Incidental Lewy bodies seen at neuropathologic examination in individuals without PD symptoms or signs are thought to represent presymptomatic individuals who would have ultimately developed clinical manifestations of PD. Lewy bodies and Lewy neurites (intra-axonal aggregates) contain the protein, α-synuclein, and are present both in PD and dementia with Lewy bodies, which is associated with wide spread cortical pathology. Other synucleinopathies include multiple system atrophy (MSA) with oligodendrocyte inclusions containing α-synuclein and some forms of neurodegeneration with brain iron accumulation (NBIA), which shows axonal spheroids with α-synuclein deposition.
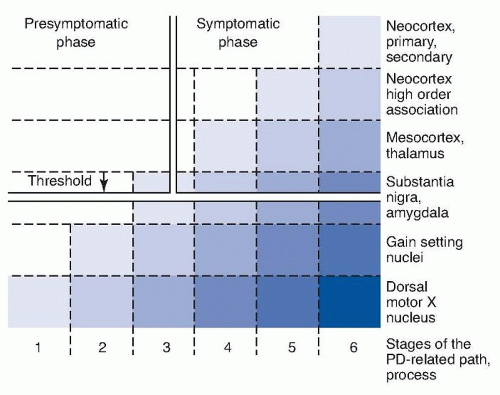
Staining for Lewy bodies and neurites with antibodies to α-synuclein indicates that the aggregation of α-synuclein first occurs in the olfactory apparatus and in the caudal brain stem, especially the dorsal motor nucleus of the vagus in the medulla even in cases without involvement of substantia nigra pars compacta or clinical PD symptoms. Braak proposed a staging system for PD pathology and proposed a hypothesis that the Lewy-related pathology progressively spread rostrally up the brain stem and then into the telencephalon and cerebral cortex (Fig. 83.1). However, Lewy pathology does not correlate with actual cell loss or function and there are many cases that do not fit the pattern of hypothesized progression. The susceptible neurons containing Lewy neurites belong to the class of projection neurons with an axon that is disproportionately long, thin, and poorly myelinated or unmyelinated.
TABLE 83.1 Classification of Major Parkinsonian Syndromes
Neurodegeneration with brain iron accumulation (NBIA)
Huntington disease (Westphal variant)
Lubag (X-linked dystonia-parkinsonism)
Mitochondrial cytopathies with striatal necrosis
Neuroacanthocytosis
Wilson disease
Parkinsonism with Neurotransmitter Enzyme Deficit
Enzymatic deficiencies of dopamine synthesis
Mn, manganese; CO, carbon monoxide; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; ALS, amyotrophic lateral sclerosis; MSA-P, multiple system atrophy with predominant parkinsonism; MSA-C, multiple system atrophy with cerebellar features.
FIGURE 83.1 Six stages of PD based on distribution of Lewy neurites in the brain. Staining for α-synuclein in Lewy neurites reveals their distribution. The darkest color represents the most intense deposition of Lewy neurites and their earliest appearance, which is in the olfactory tubercle and dorsal motor nucleus of the vagus. The substantia nigra develops Lewy neurites at stage 3, following which symptoms of PD appear (Braak stage 4). The neocortex is involved in stages 5 and 6. The susceptible neurons belong to the class of projection neurons with an axon that is disproportionately long, thin, and poorly myelinated or unmyelinated. (From Braak H, Bohl JR, Müller CM, et al. Stanley Fahn lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21[12]:2042-2051.)
ETIOLOGY
The cause of PD in the vast majority of patients is unknown. Research has discovered both environmental and genetic factors. The discovery that the chemical agent 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can cause parkinsonism raised the possibility that PD might be caused by an environmental toxin. No single environmental factor has emerged as essential, but growing up in a rural farming environment has been disproportionately frequent in some studies, suggesting potential role of pesticides, some resembling MPTP in chemical structures. Exposure to excess levels of manganese may produce a neurobehavioral syndrome that shares some of the cognitive and motor features of PD, but whether it increases the risk of idiopathic PD is controversial. Interestingly, decreased risk of PD in those with high level of caffeine consumption or cigarette smoking has been consistently noted. High uric acid levels are also associated with decreased risk and slower progression of PD.
GENETICS
Contribution of genetic factors has been recognized only since late 1990s. Studies of twins showed that onset of PD before age 50 years has a higher likelihood of a genetic cause. The first genetic form of PD was discovered in 1997 and now more than 20 different genetic forms of parkinsonism, labeled as PARK, have been discovered. Many of them are Parkinson-plus syndromes, and pathology is variable including some without Lewy body and some as NBIAs. These are summarized in Table 83.2. We will discuss wellestablished genetic forms with typical PD clinical phenotypes here.
TABLE 83.2 The Most Definitive Genetic Forms of Parkinson Disease
Name and Locus
Gene
Mode of Inheritance
Pathologic and Clinical Features
Protein Function
Where Found
PARK1 and 4 4q22.1
SCNA
Autosomal dominant
Lewy bodies; earlier onset and more aggressive course; L-dopa-responsive parkinsonism, dementia, hallucinations, autonomic dysfunction
α-Synuclein possibly synaptic vesicle trafficking, elevated in bird song learning
Families in Germany, Italy, United States (Contoursi kindred), Greece, Spain
PARK8 12q12
LRRK2
Autosomal dominant
Pathologic pleomorphism; indistinguishable from idiopathic PD
Worldwide
PARK2 6q26
PRKN
Autosomal recessive
Often juvenile onset without Lewy bodies; slowly progressive; no dementia
Parkin, a ubiquitin E3 ligase, attaches short ubiquitin peptide chains to a range of proteins, likely to mark degradation; supports mitophagy
Ubiquitous, originally in Japan, very common in juvenile onset
Families in Italy, Spain, Philippines, Taiwan, Israel, Japan, Ireland, and North America
PARK7 1p36.23
DJ-1
Autosomal recessive
Early onset
Possible atypical peroxiredoxin and may play a role in apoptosis
Families in Holland, Italy, Uruguay
Glucocerebrosidase 1q22
GBA
Autosomal dominant; susceptibility gene; low penetrance; most common genetic risk factor gene
Indistinguishable from idiopathic PD
Lysosomal enzyme
About 13% of sporadic PD in Ashkenazi Jews, found in all ethnic groups
PD, Parkinson disease.
The first genetic cause of PD (PARK1) was discovered to be mutations in SNCA gene located on chromosome 4q22.1 coding for the protein α-synuclein. α-Synuclein is an abundant presynaptic protein. The resulting parkinsonism transmits in an autosomal dominant pattern. It is rare, being reported only in a small number of families originating in Greece, Italy, Germany, and Spain. However, single nucleotide polymorphisms have been associated with sporadic PD and the protein α-synuclein is present in Lewy bodies, even in patients with PD without genetic mutations, suggesting potential pathophysiologic role of α-synuclein in sporadic form of PD. It is not known if Lewy bodies contribute to neuronal degeneration or are a protective mechanism to slow neuronal death. Duplication and triplication of the α-synuclein gene also cause familial parkinsonism (PARK4), suggesting that overexpression of the normal (wild-type) protein suffices to provoke dopaminergic neurodegeneration.
Another gene defect causing familial PD is PARK8, which has been mapped to chromosome 12q12 and encodes for a previously unknown protein named leucine-rich repeat kinase- 2 (LRRK2), also known as dardarin. This 2527 amino acid protein belongs to the family of the ROCO protein, contains a protein kinase domain, and is ubiquitously expressed in the central nervous system (CNS). About a dozen pathogenic LRRK2 mutations have been identified, which have been found to be the most frequent genetic cause of PD, accounting to up to 5% of familial cases in the Caucasian population. Some ethnic groups have a particularly high prevalence; the most common mutation, G2019S, has an increased frequency among Ashkenazi Jews (18.3% of those with PD) and North African Berbers (39% of those with PD). LRRK2 mutations result in an autosomal dominant parkinsonism that resembles late-onset idiopathic PD. Genome-wide association studies have shown significance of LRRK2 polymorphism in sporadic PD as well. Although the neuropathology associated with LRRK2 mutations is highly variable with some with Lewy bodies and others with neurofibrillary tangles, degeneration of substantia nigra neurons has been observed consistently.
The most commonly occurring gene defect causing juvenile familial parkinsonism is PARK2 (PRKN) on chromosome 6q26, coding for the E2-dependent E3 ubiquitin-protein ligase, parkin. Mutations in the parkin gene result in an autosomal recessive parkinsonism that is slowly progressive, with onset usually before age 30 years and with sleep benefit; rest tremor can be present. This cause of PD has a better prognosis for both motor and cognitive outcomes than idiopathic PD. There is degeneration of substantia nigra neurons, but in most instances, no Lewy body inclusions are found. Some typical adult-onset PD patients have been found to have a single heterozygotic mutation of the PRKN gene and with Lewy bodies at autopsy. Other recessive forms with similar juvenile parkinsonism include those with PTEN-induced putative kinase 1 (PINK1; PARK6) and DJ-1 (PARK7) mutations.
Glucocerebrosidase (GBA) gene mutations, when homozygous, cause autosomal recessive Gaucher disease. Heterozygous carriers are at increased risk for developing PD that is indistinguishable from idiopathic PD. PD patients with GBA mutations tend to have more cognitive problems, and about 13% of Ashkenazi Jews with PD have this mutation, but the same mutation causes PD in other ethnic groups as well.
PATHOGENESIS
Two major pathogenic hypotheses have emerged from epidemiologic and genetic evidences as well as postmortem examinations. One hypothesis proposes that mitochondrial dysfunction and oxidative stress are critical in the pathogenesis, whereas there is also evidence that misfolding and aggregation of proteins are instrumental in the PD neurodegenerative process. These two hypotheses are not mutually exclusive, and interactions among these pathogenic factors are likely to be important in understanding the mechanisms of neurodegeneration in PD.
Environmental toxins associated with PD risk can damage mitochondria and generate oxidative stress. Postmortem biochemical observations also show that complex I activity of mitochondria is reduced in substantia nigra of patients with PD. Such a defect would decrease the synthesis of ATP and also lead to the buildup of free electrons, thereby increasing oxidative stress. Substantia nigra in patients with PD shows severe depletion of reduced glutathione, the major substrate required for the elimination of reactive oxygen species. This change is also seen in brains with incidental Lewy bodies and therefore could be the one of the earliest biochemical abnormalities of PD. It is not known, however, if this change is the cause or the result of oxidative stress. Iron in the substantia nigra may also play a critical role because it can catalyze the formation of the highly reactive hydroxyl radical from hydrogen peroxide. Recessive genes such as PRKN and PINK1 have been shown to have wide ranging effects on mitochondrial quality control such as regulating mitochondria biogenesis, maintaining fission-fusion balance to remove damaged mitochondria, transport of mitochondria, and turnover of damaged mitochondria by recruiting autophagic machinery. Another recessive gene, DJ-1 is implicated in a pathway handling oxidative stress. Endogenous factors may also predispose melanin-containing monoaminergic neurons to neurodegeneration. Cellular oxidation reactions (such as enzymatic oxidation and auto-oxidation of dopamine and other monoamines) result in the formation of reactive oxygen species such as dopamine quinone and other metabolic products that can damage the monoamine neurons. In addition, dopaminergic neurons and other susceptible neurons such as noradrenergic neurons in locus ceruleus are autonomous pacemakers and have long highly arborized axons, thereby subjected to high metabolic demands. A presence of a particular calcium channel may predispose these neurons to basal metabolic stress.
Most of the genes involved in PD seem to have multiple cellular functions. Nonetheless, genes producing recessive forms of PD have been implicated more in mitochondrial and metabolic pathways as noted earlier, whereas genes associated with autosomal dominant forms of PD have been noted to be involved in protein degradation homeostasis. Whether the hallmark of PD pathology, the Lewy body, which contains α-synuclein contributes to the toxicity or is a protective mechanism is not known. Evidence points to toxicity of protofibrillar forms of α-synuclein, and intriguing experimental evidence suggests that abnormal forms of α-synuclein may have the prion-like property of propagating its abnormal conformation and spreading the pathology along the neuronal projections and across the synaptic connections. α-Synuclein is involved in vesicle recycling. LKKR2 plays a role in vesicular trafficking and cytoskeletal function. Another PD gene, vacuolar protein sorting 35 (VPS35) was identified to cause autosomal dominant PD with typical features and good response to levodopa. This gene encodes a subunit of the retromer complex involved in endosomes and vesicular recycling. α-Synuclein and LRRK2 affects protein degradation pathways, including autophagy, a process of cell degradation of dysfunctional cellular components by lysosomes. Glucocerebrosidase is a lysosomal enzyme.
CLINICAL MANIFESTATIONS
CARDINAL MOTOR FEATURES
The clinical features of tremor, bradykinesia, rigidity, loss of postural reflexes, flexed posture, and freezing are the six cardinal motor features of parkinsonism. Not all need to be present, but at least two should be seen, either rest tremor or bradykinesia, before parkinsonism is considered clinically probable. Rest tremor at a frequency of 4 to 5 Hz is present in the extremities, almost always distally; occasionally, the rest tremor occurs in the proximal part of the limb instead of distally. The classic “pill-rolling” tremor involves the thumb and forefinger. Rest tremor disappears with action but sometimes reemerges as the limbs maintain a posture (Video 83.1). Rest tremor is also common in the lips, chin, and tongue. Rest tremor of the hands increases with walking and may be an early sign when others are not yet present. Stress or excitement worsens the tremor. Some patients will have an action tremor instead of or in addition to rest tremor. The biggest differential diagnosis of parkinsonian tremor is essential tremor (ET). Ordinarily, these two causes of tremor are easy to distinguish. Tremor of PD is typically a rest tremor, whereas that of ET is a postural and action tremor. However, the manifestations of these tremors can overlap, with patients with severe ET having rest tremor. Not all patients with PD have tremor; when absent, the diagnosis is more difficult to make, and it usually takes longer after onset of symptoms to make a diagnosis. Eventually, bradykinesia worsens to the point of some disability, such as micrographia or dragging a leg when walking, that the patient seeks medical attention.
Akinesia is a paucity of spontaneous movement, but the term is often used interchangeably with bradykinesia and hypokinesia. Bradykinesia (slowness of movement, difficulty initiating movement, and loss of automatic movement) and hypokinesia (reduction in amplitude of movement, particularly with repetitive movements, so-called decrementing) are the most common features of parkinsonism, although they may appear after the tremor. Bradykinesia has many facets, depending on the affected body parts. The face loses spontaneous expression (masked facies, hypomimia) with decreased frequency of blinking. Poverty of spontaneous movement is characterized by loss of gesturing and by the patient’s tendency to sit motionless. Speech becomes soft (hypophonia), and the voice has a monotonous tone with a lack of inflection (aprosody). Some patients do not enunciate clearly (dysarthria) and do not separate syllables clearly, thus running the words together (tachyphemia). Bradykinesia of the dominant hand results in small and slow handwriting (micrographia) and in difficulty shaving, brushing teeth, combing hair, buttoning, or applying makeup. Playing musical instruments is impaired. Walking is slow, with a shortened stride length and a tendency to shuffle with loss of heel strike; arm swing decreases and eventually is lost. Difficulty arising from a deep chair, getting out of automobiles, and turning in bed are symptoms of truncal/body bradykinesia. Drooling saliva results from failure to swallow spontaneously, a feature of bradykinesia, and is not caused by excessive production of saliva. The patients can swallow properly when asked to do so but only constant reminders allow them to keep swallowing saliva. Similarly, arm swing can be normal if the patient voluntarily, and with effort, wishes to have the arms swing on walking. Pronounced bradykinesia prevents a patient with parkinsonism from driving an automobile when foot movement from the accelerator to the brake pedal is too slow.
Bradykinesia is commonly misinterpreted by patients as weakness. Fatigue, a common complaint in PD, particularly in the mild stage of the disease before pronounced slowness appears, may be related to mild bradykinesia or rigidity or may be an independent feature. Subtle signs of bradykinesia can be detected even in the early stage of parkinsonism if one examines for slowness in shrugging the shoulders, lack of gesturing, decreased arm swing, and decrementing amplitude of rapid successive movements. With advancing bradykinesia, slowness and difficulty in the execution of activities of daily living increase. A meal normally consumed in 20 minutes may be only half eaten in an hour or more. Swallowing may become impaired with advancing disease, and choking and aspiration are concerns. Bradykinesia is manifested in many ways depending on the body part affected (Table 83.3).
Rigidity is an increase in muscle tone that is elicited when the examiner moves the patient’s limbs, neck, or trunk. This increased resistance to passive movement is equal in all directions and usually is manifested by a ratchety “give” during the movement. This so-called cogwheeling is caused by the underlying tremor even in the absence of visible tremor. Cogwheeling also occurs in patients with ET. Rigidity of the passive limb increases while another limb is engaged in voluntary active movement (“Froment maneuver”).
Flexed posture commonly begins in the arms and spreads to involve the entire body. The head is bowed; the trunk is bent forward; the back is kyphotic; the arms are held in front of the body; and the elbows, hips, and knees are flexed. Deformities of the hands include ulnar deviation of the hands, flexion of the metacarpophalangeal joints, and extension of the interphalangeal joints (striatal hand). Inversion of the feet is apparent, and the big toes may be dorsiflexed (striatal toe) and the other toes curled downward. Lateral tilting of the trunk commonly develops (Pisa syndrome), and extreme flexion of the trunk (camptocormia) is sometimes seen.
Loss of postural reflexes leads to falling and eventually to inability to stand unassisted. Postural reflexes are tested by the pull test, which is performed by the examiner, who stands behind the patient and gives a sudden firm pull on the shoulders after explaining the procedure and who checks for retropulsion (Videos 83.2 and 83.3). With advance warning, a normal person can recover within two steps. The examiner should always be prepared to catch the patient when this test is conducted; otherwise, a person who has lost postural reflexes could fall. The examiner should have a solid wall behind him or her in case a heavy patient falling backward also causes the examiner to fall backward. As postural reflexes are impaired, the patient collapses into the chair on attempting to sit down (sitting en bloc). Walking can be marked by festination, whereby the patient walks faster and faster, trying to move the feet forward to be under the flexed body’s center of gravity and thus prevent falling.
The freezing phenomenon (motor block) is a transient inability to perform active movements. It most often affects the legs when walking but also can involve eyelid opening (known as apraxia of lid opening or levator inhibition), speaking (palilalia), and writing. Freezing occurs suddenly and is transient, lasting usually no more than several seconds with each occurrence. The feet seem as if “glued to the ground” and then suddenly become “unstuck,” allowing the patient to walk again (Video 83.3). Freezing typically occurs when the patient begins to walk (start hesitation); turns while walking; or approaches a destination, such as a chair in which to sit (destination hesitation); it is often induced when the patient walks in crowded places (e.g., in the narrow confines of a theater row or when trying to go through a revolving door, or when suddenly confronted by a person coming into their path) and when there is a time restriction to the walking (e.g., trying to enter or exit an elevator before the door closes or when trying to cross a street before the traffic light turns to red). Freezing is often overcome by visual clues, such as having the patient step over objects, and is much less frequent when the patient is going up or down steps than when walking on level ground. The combination of freezing of gait and loss of postural reflexes is particularly devastating because it often leads to falls. Falling is responsible for the high incidence of hip fractures in parkinsonian patients.
TABLE 83.3 Clinical Signs of Brradykinesia
Cranial
Hypomimia (masked face)
Staring expression with decreased blinking and retracted lids
Hypometric saccades
Impaired convergence
Impaired upward gaze
Hypophonia (soft voice)
Aprosody of speech (loss of inflection of voice)
Palilalia (repetition of first syllable)
Sialorrhea (drooling of saliva)
Upper Limbs
Reduced spontaneous movement (e.g., lack of gesturing)
Decrementing amplitude with repetitive movements of opening and closing fists, pronating and supinating the forearms, and tapping a finger on the thumb
Micrographia and slowness with handwriting
Slowness in cutting food, dressing, and in hygienic care
Decreased arm swing when walking
Lower Limbs
Decrementing amplitude with repetitive movements of stomping feet or tapping toes
Short, slow steps when walking
Not elevating feet as high as normal when walking, tendency to shuffle
Narrow base when walking
Tendency to walk on toes; loss of heel strike
Body Bradykinesia
Slowness in initiating movement on command
Difficulty arising from a chair and turning in bed
Difficulty carrying out two motor acts simultaneously
Reduced shrugging of shoulders
SYMPTOMS AND SIGNS
The clinical signs and symptoms of PD can be divided into motor and nonmotor features of PD and those due to complications or adverse effects of the medications employed to treat the disease. The clinical motor features of PD are represented within the six cardinal features of parkinsonism discussed earlier. Of the six cardinal motor signs, tremor, bradykinesia, and rigidity occur early in the course of the disease, whereas flexed posture, loss of postural reflexes, and freezing of gait occur in more advanced stages. Falling is a late symptom. If these normally advanced signs and symptoms occur within 2 years of onset, one should suspect another cause of parkinsonism, such as progressive supranuclear palsy or MSA.
The onset of PD is insidious; tremor is the symptom first recognized in 70% of patients (Table 83.4). Symptoms often begin unilaterally; as the disease progresses, symptoms and signs become bilateral. The disease can remain confined to one side for several years before the other side becomes involved. The disease progresses slowly, and if untreated, the patient eventually becomes wheelchairbound and bedridden. Despite having severe bradykinesia with marked immobility, patients, when presented with a sudden stimulus, may rise suddenly and move normally for a short burst of motor activity, so-called kinesia paradoxica. The Hoehn-Yahr clinical staging (Table 83.5) captures the progression of the motor features of PD, from unilateral to bilateral, to loss of postural reflexes to disability. Disability in carrying out activities of daily living is scored on the Schwab-England scale. More detailed scoring of individual signs and symptoms of PD are captured in the Unified Parkinson’s Disease Rating Scale (UPDRS) and its newer version that includes more nonmotor symptoms, the Movement Disorder Society (MDS)-UPDRS.
TABLE 83.4 Initial Symptoms in Parkinson Disease
Symptoms
No. of Cases (n = 183)
Percentage
Tremor
129
70.5
Stiffness or slowness of movement
36
19.7
Loss of dexterity and/or handwriting disturbance
23
12.6
Gait disturbance
21
11.5
Muscle pain, cramps, aching
15
8.2
Depression, nervousness, or other psychiatric disturbance
8
4.4
Speech disturbance
7
3.8
General fatigue, muscle weakness
5
2.7
Drooling
3
1.6
Loss of arm swing
3
1.6
Facial masking
3
1.6
Dysphagia
1
0.5
Paresthesia
1
0.5
Average number of initial symptoms per patient
1.4
TABLE 83.5 The Modified Hoehn and Yahr Staging Scale for Parkinson Disease
Stage 0
No signs of disease
Stage 1
Unilateral disease
Stage 1.5
Unilateral plus midline/axial involvement
Stage 2
Bilateral disease, without impairment of balance
Stage 2.5
Mild bilateral disease, with an abnormal pull test but with recovery to avoid falling
Stage 3
Mild to moderate bilateral disease; some postural instability (would fall on the pull test if not caught); physically independent
Stage 4
Severe disability; still able to walk or stand unassisted, but a walking aid is advisable to prevent falling
Stage 5
Wheelchair-bound or bedridden unless aided
From Fahn S, Elton RL, Members of the UPDRS Development Committee. The unified Parkinson’s disease rating scale. In Fahn S, Marsden CD, Calne DB, et al, eds. Recent Developments in Parkinson’s Disease. Vol. 2. Florham Park, NJ: Macmillan Healthcare Information; 1987:153-163, 293-304.
The nonmotor symptoms of PD (Table 83.6) can be more troublesome than the motor features of PD. Behavioral and personality changes include a reduced attention span, visuospatial impairment, and a personality that slowly becomes more dependent, fearful, indecisive, and passive. The spouse gradually makes more of the decisions and becomes the dominant partner. The patient speaks less spontaneously. The patient eventually sits much of the day and is inactive unless encouraged to exercise. Passivity and lack of motivation are common and are expressed by the patient’s aversion to visiting friends. The patient is more reticent to participate in conversations. Depression is frequent in patients with PD, with about 25% to 50% prevalence. Anxiety may be even more common, often with depression.
Cognitive decline is not an early feature but can become pronounced as the patient ages. Memory impairment, in contrast to AD, is not a feature of early PD; rather, the patient is just slow in responding to questions, so-called bradyphrenia. The correct answer can be obtained if the patient is given enough time. Subtle signs of bradyphrenia include tip-of-the-tongue phenomena from diminished verbal fluency and the inability to change mental set rapidly. In a cross section of patients with PD, 15% to 20% have a more profound dementia, but as many as 75% will develop dementia in their late 70s. Most of these patients have developed Lewy bodies in cortical neurons (Parkinson disease dementia [PDD]), and some have developed concurrent AD. These disorders are not always distinguishable, but dementia with Lewy bodies is often characterized by fluctuating hallucinations.
Sensory symptoms are fairly common, but objective sensory impairment is not seen in PD. Symptoms of pain, burning, and tingling occur in the region of motor involvement. A patient may have dull pain in one shoulder as an early symptom of the disease, which often is misdiagnosed as arthritis or bursitis, and even before clearcut signs of bradykinesia appear in that same arm. Akathisia (inability to sit still, restlessness) and the restless legs syndrome (RLS) occur in some patients with PD. In both syndromes, uncomfortable sensations disappear with movement, and sometimes, the two conditions are difficult to distinguish. Akathisia, if present, is usually present most of the day; it may respond to levodopa but otherwise has not been treated successfully. The RLS develops late in the day with crawling sensations in the legs and may be associated with periodic movements in sleep, thereby disturbing sleep. Other sleep problems are fragmented sleep and rapid eye movement (REM) sleep behavior disorder (acting out one’s dreams); the latter is usually successfully treated with clonazepam; melatonin may also provide relief. Other sleep problems are encountered with dopaminergic medications including excessive daytime sleepiness and sudden attacks of sleep without warning.
TABLE 83.6 Nonmotor Features of Parkinson Disease
Personality and Behavior
Depression
Fearfulness
Anxiety
Loss of assertive drive
Passivity
Greater dependence
Inability to make decisions
Loss of motivation, apathy
Abulia
Cognition and Mental State
Bradyphrenia
“Tip of the tongue” phenomenon
Confusion
Dementia
Sleep Problems
Sleep fragmentation
REM sleep behavior disorder
Excessive daytime sleepiness
Altered sleep-wake cycle
Drug-induced sleep attacks
Sensory
Pain
Paresthesia, numbness
Burning
Akathisia
Restless legs syndrome
Hyposmia
Autonomic
Orthostatic hypotension
Bladder problems
Gastrointestinal; constipation
Sexual dysfunction
Seborrhea
Sweating
Rhinorrhea
Behavioral Problems due to Medications
Hallucinations
Psychosis
Punding
Compulsive behaviors
Nonmotor offs
Other
Fatigue
REM, rapid eye movement.
Only gold members can continue reading. Log In or Register to continue