Fig. 17.1
Small cell carcinomas are characterized by loosely arranged sheets of small cells with a high N:C ratio, hyperchromatic and finely granular chromatin, inconspicuous nucleoli, and nuclear molding. High mitotic activity and necrosis are also present
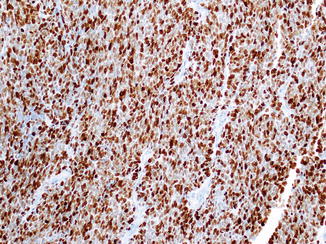
Fig. 17.2
Ki-67 labeling index of poorly differentiated neuroendocrine carcinomas is much higher than the WHO-recommended threshold (>20 %) for the grade 3 category. A small cell carcinoma with >95 % Ki-67 labeling index is depicted here
The large cell variant (large cell neuroendocrine carcinoma) is more common and characterized by large cells with prominent nucleoli and variable amounts of cytoplasm. Diffuse, nested, trabecular, gland-forming, and peripheral palisading growth patterns, often intermingled in varying proportions, are seen in most large cell neuroendocrine carcinomas (Fig. 17.3). Some tumors display pseudopapillae, composed of viable tumor cells surrounding fibrovascular cores, usually at the periphery of necrotic areas. Apoptotic cells and mitotic figures are abundant, but mitotic figures in the large cell neuroendocrine carcinomas are usually not as numerous as in the small cell carcinomas. In the aforementioned study, the average mitotic count and Ki-67 labeling index of large cell neuroendocrine carcinomas were found to be 37 per 10 high power fields and 66 %, respectively [3].
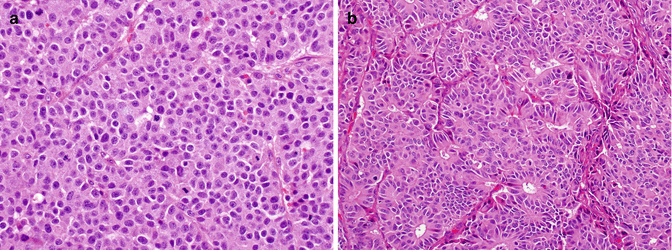
Fig. 17.3
Large cell neuroendocrine carcinoma may reveal various growth patterns (a, trabecular; b, gland-forming pattern). Compared to small cell carcinoma, their cells are larger and round to polygonal with round nuclei that have vesicular chromatin or prominent nucleoli
In cases with the typical cytologic features of small cell carcinoma, it is not necessary to document neuroendocrine differentiation by immunohistochemistry, provided alternative diagnoses (primitive neuroectodermal tumor, desmoplastic small round cell tumor, etc.) can be excluded. However, for large cell neuroendocrine carcinomas, positive immunohistochemical staining for chromogranin or synaptophysin should be obtained to confirm the diagnosis [1, 6, 15], although the extent and intensity of staining are usually less than in well-differentiated NETs of the pancreas.
Some PD-NECs may be associated with exocrine pancreatic neoplasm component, in the form of acinar cell carcinoma (Fig. 17.4) or in the form of ductal adenocarcinoma (Fig. 17.5), intraductal papillary mucinous neoplasm, or even squamous cell carcinoma [1, 3, 6, 16–19]. The 2010 WHO classification system recommends that at least 30 % of either component be present in order to qualify a tumor as “mixed carcinoma” [1], although the figure of 25 % has also been advocated in the AFIP fascicle on tumors of the pancreas [6].
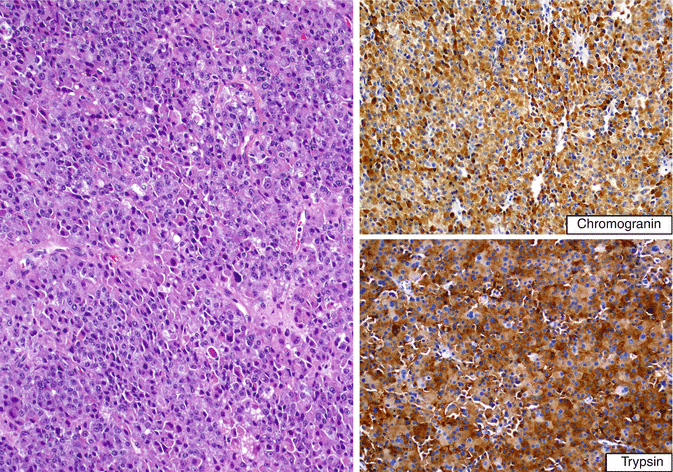
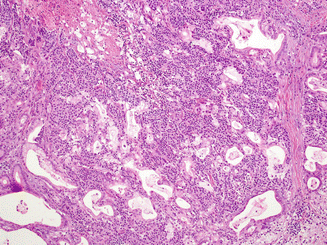
Fig. 17.4
Mixed acinar-neuroendocrine carcinomas are usually composed of morphologically homogenous population of cells, and the divergent differentiation cannot be detected without immunohistochemical labeling for both neuroendocrine and acinar differentiation markers (chromogranin and trypsin immunohistochemical stains are depicted here)
Fig. 17.5
In contrast to mixed acinar-neuroendocrine carcinomas, different components are morphologically recognizable in mixed ductal-neuroendocrine carcinomas, even if they are intimately admixed as illustrated here, as gland formations and presence of abundant intracellular mucin are striking
In mixed ductal- or squamous-neuroendocrine carcinomas, different components, sharply segregated or intimately admixed, are usually morphologically recognizable (Fig. 17.4). Of note, PD-NECs, especially large cell neuroendocrine carcinomas, with gland formations should not be mistaken for mixed ductal-neuroendocrine carcinomas. Absence of detectable mucin and careful attention to the cytologic appearance aid in proper diagnosis in the former (see Chap. 18 for detailed discussion). In contrast, mixed acinar-neuroendocrine carcinomas display less segregation of the two cell types (Fig. 17.4). In fact, in most cases, it is not possible to recognize with certainty that two lines of differentiation are present without immunohistochemical staining for both neuroendocrine and acinar differentiation markers [3, 6, 19]. If only neuroendocrine staining is performed, these cases can be misinterpreted as neuroendocrine carcinomas. Therefore, it is not surprising that many cases that are diagnosed as PD-NECs prove to be mixed acinar-neuroendocrine carcinoma once studied more carefully [3, 19].
On cytology, small cell carcinoma is characterized by often crushed, small- to intermediate-sized tumor cells with irregular nuclear borders, high nucleus-to-cytoplasm ratio, and nuclear molding. In addition to brisk mitotic activity, there is extensive single-cell and background necrosis. For large cell neuroendocrine carcinoma, tumor cells are usually larger with variable cytoplasm as well as prominent central nucleoli. However, these tumors may resemble a poorly differentiated adenocarcinoma and may not show the classical coarsely clumped chromatin of well-differentiated NETs or the nuclear molding of their small cell counterpart. If a neuroendocrine carcinoma is not suspected at the time of evaluation, the diagnosis may be easily missed [20].
17.4 Differential Diagnosis
Primary pancreatic PD-NECs are extremely rare and must be distinguished from metastatic PD-NECs from another organ or direct invasion from a contiguous site, particularly the ampulla of Vater [21] or stomach. When there is a single mass in the pancreas and, after critical evaluation, no convincing clinical, radiographic, or pathologic evidence of a lung primary (or another site), then pancreatic origin is reasonable. It should be kept in mind that TTF1 immunohistochemical staining is not helpful, as small cell carcinomas from both pulmonary and a variety of extrapulmonary sites are TTF1 positive. Clinical information and a history of a previous carcinoma are especially important in the accurate diagnosis of such cases.
Pancreatic well-differentiated NETs can show significant nuclear pleomorphism or small cell change (high nuclear/cytoplasmic ratio resembling small cell carcinoma) [22], and features such as a markedly infiltrative growth pattern and necrosis can also be identified [6]. On casual examination, these findings falsely suggest a PD-NEC. If thorough mitotic counting as well as immunolabeling for Ki-67 are not performed, misclassification can occur, which can have therapeutic consequences [2, 23]. A recent landmark study has shown that not all patients with a grade 3 NEC defined based on WHO 2010 criteria benefit from the platinum-based chemotherapy typically used for PD-NECs. In this study, grade 3 tumors with a Ki-67 index <55 % were less responsive than grade 3 NECs with a Ki-67 index ≥55 %, although the latter group experienced early recurrence of shorter ultimate survival than the group with a Ki-67 in the 20–55 % range [2], supporting the concept that the 2010 WHO grade 3 category is heterogeneous, and the tumors at the lower end of the grade 3 range are in fact well-differentiated NETs with an elevated proliferation rate. These well-differentiated NETs typically have a Ki-67 index around 40 %, whereas PD-NECs’ Ki-67 index is about 70 % [3, 4]. Also, a recent study has shown that most pancreatic PD-NECs abnormally immunolabel for p53 (nuclear expression) and Rb (loss of expression) in 95 % and 74 % of cases, respectively. In that study, the abnormal expression of these proteins correlated with intragenic mutations in the TP53 and retinoblastoma genes coding for these proteins. By contrast, immunohistochemical studies only detect rare p53 abnormalities and Rb immunolabeling is intact in pancreatic well-differentiated NETs [5, 24]. In addition, approximately 45 % of sporadic pancreatic well-differentiated NETs show mutually exclusive loss of expression of DAXX (death-domain-associated protein) or ATRX (α-thalassemia/mental retardation syndrome X-linked) immunohistochemical stains, which correlates with mutations in the DAXX and ATRX genes [5, 25] (see Molecular Pathology section for more details).
Related posts:
 Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
Endocrinological Approach to the Diagnosis of Pancreatic Neuroendocrine Neoplasms
Molecular Pathology of Pancreatic Neuroendocrine Neoplasms
 Nonfunctioning Pancreatic Neuroendocrine Neoplasms (Including PP-Producing and Calcitonin-Producing Tumors)
Nonfunctioning Pancreatic Neuroendocrine Neoplasms (Including PP-Producing and Calcitonin-Producing Tumors)
 Insulinoma
Insulinoma
 Hyperplastic and Microadenomatous Pancreatic Neuroendocrine Lesions
Hyperplastic and Microadenomatous Pancreatic Neuroendocrine Lesions
 Classification and Staging of Pancreatic Neuroendocrine Neoplasms
Classification and Staging of Pancreatic Neuroendocrine Neoplasms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


