Primary Central Nervous System Lymphoma
Tracy T. Batchelor
INTRODUCTION
Primary central nervous system lymphoma (PCNSL) is an extranodal non-Hodgkin lymphoma confined to the brain, leptomeninges, eyes, or spinal cord. The prognosis of PCNSL is inferior to that of other non-Hodgkin lymphoma subtypes. The diagnosis and management of PCNSL differs from that of other primary brain cancers and non-Hodgkin lymphoma in other parts of the body.
EPIDEMIOLOGY
PCNSL is a rare brain tumor and subtype of non-Hodgkin lymphoma. An estimated 3,855 cases of PCNSL were diagnosed in the United States from 2004 to 2006, and the number of cases is expected to increase further with the aging of the U.S. population. The median age at diagnosis is 65 years and PCNSL is slightly more common among men. PCNSL accounts for approximately 3% of all the primary central nervous system (CNS) tumors diagnosed each year in the United States. Between 1970 and 2000, the incidence of PCNSL increased, largely due to the HIV pandemic. The annual incidence rate is 0.47 cases per 100,000 person-years. Since 2000, there has been a further increase in the incidence of PCNSL, especially in the elderly. Congenital or acquired immunodeficiency is the only established risk factor for PCNSL and individuals infected with the HIV are at greater risk of developing this tumor. However, the incidence of HIV-related PCNSL has declined dramatically, and this chapter focuses on PCNSL in the immunocompetent host.
PATHOBIOLOGY
Approximately 90% of PCNSL cases are diffuse large B-cell lymphomas (DLBCL), with the remainder consisting of T-cell lymphomas, poorly characterized low-grade lymphomas, or Burkitt lymphomas. Primary CNS DLBCL is composed of centroblasts or immunoblasts clustered in the perivascular space, with reactive lymphocytes, macrophages, and activated microglial cells intermixed with the tumor cells. Most tumors express pan-B-cell markers including CD19, CD20, CD22, and CD79a. The molecular mechanisms underlying transformation and localization to the CNS are poorly understood. Limitations in molecular studies of PCNSL include the rarity of the disease and the limited availability of tissue because the diagnosis is most often made with stereotactic needle biopsy. Like systemic DLBCL, PCNSL harbors chromosomal translocations of the BCL6 gene, deletions in 6q, and aberrant somatic hypermutation in protooncogenes including MYC and PAX5. Inactivation of CDKN2A is also commonly observed in both entities. Also like DLBCL, PCNSL can be classified into the three molecular subclasses by gene expression profiling: type 3 large B-cell lymphoma, germinal center B-cell (GCB) lymphoma, and activated B-cell lymphoma (ABC). In all DLBCL cases, the ABC gene expression profile is associated with an inferior prognosis versus the GCB profile. The ABC subclass accounts for a higher proportion of primary CNS DLBCL cases versus other types of DLBCL. This higher prevalence of the ABC gene expression profile subtype in PCNSL may partially account for the relatively inferior prognosis of this lymphoma versus other forms of DLBCL. Moreover, certain molecular features distinguish primary CNS DLBCL from systemic DLBCL. Gene expression profiles demonstrate that PCNSL is characterized by differential expression of genes related to adhesion and the extracellular matrix pathways, including MUM1, CXCL13, and CHI3L1. The ongoing somatic hypermutation with biased use of VH gene segments that has been observed in PCNSL is suggestive of an antigen-dependent proliferation. These observations are consistent with the hypothesis that PCNSL is secondary to antigen-dependent activation of circulating B cells, which subsequently localize to the CNS by expression of various adhesion and extracellular matrix-related genes. However, further molecular studies to investigate the transforming events and the subsequent events responsible for CNS tropism in PCNSL are needed. Insights into the molecular pathogenesis of PCNSL may allow the development of targeted therapeutic approaches for this tumor.
CLINICAL MANIFESTATIONS
The median age of immunocompetent patients diagnosed with PCNSL is 60 years. The presenting symptoms and signs are variable. In 248 immunocompetent patients, 43% had neuropsychiatric signs, 33% had symptoms of increased intracranial pressure, 14% had seizures, and 4% had ocular symptoms. Seizures are less common than with other types of brain tumors probably because PCNSL involves predominantly subcortical white matter rather than epileptogenic gray matter. Unlike patients with systemic non-Hodgkin lymphoma, PCNSL patients rarely manifest B symptoms.
DIAGNOSIS
The International PCNSL Collaborative Group (IPCG) has developed guidelines to determine extent of disease. A gadoliniumenhanced brain magnetic resonance imaging (MRI) scan is the most sensitive radiographic study for the detection of PCNSL (Fig. 99.1). Most PCNSL patients present with a single brain mass. The mass is typically isointense to hyperintense on T2-weighted MRI sequences and homogeneously enhancing on postcontrast images. The diagnosis of PCNSL is typically made by stereotactic brain biopsy, by cerebrospinal fluid (CSF) analysis, or by analysis of vitreous aspirate in patients with ocular involvement. Given the possible delay in diagnosis and treatment with the latter two methods, prompt stereotactic biopsy is advised in almost all cases that are surgically accessible. Secondary CSF and ocular involvement occurs in approximately 15% to 20% and 5% to 20% of PCNSL patients, respectively. Presenting symptoms of
ocular involvement include eye pain, blurred vision, and floaters. B symptoms such as weight loss, fevers, and night sweats are infrequent in PCNSL. A thorough diagnostic evaluation is needed to establish the extent of the lymphoma and to confirm localization to the CNS. Physical examination should consist of a lymph node examination, a testicular examination in men, and a comprehensive neurologic examination. A lumbar puncture should be performed if not contraindicated, and CSF should be assessed by flow cytometry, cytology, and immunoglobulin heavy-chain gene rearrangement. Because extraneural disease must be excluded to establish a diagnosis of PCNSL, computed tomography (CT) or CT/positron emission tomography (PET) scans of the chest, abdomen, and pelvis and a bone marrow biopsy and aspirate should be performed to exclude occult systemic disease. Involvement of the optic nerve, retina, or vitreous humor should be excluded with a comprehensive eye evaluation by an ophthalmologist that includes a slit lamp examination. Blood tests should include a complete blood count, a basic metabolic panel, serum lactate dehydrogenase, and HIV serology.
ocular involvement include eye pain, blurred vision, and floaters. B symptoms such as weight loss, fevers, and night sweats are infrequent in PCNSL. A thorough diagnostic evaluation is needed to establish the extent of the lymphoma and to confirm localization to the CNS. Physical examination should consist of a lymph node examination, a testicular examination in men, and a comprehensive neurologic examination. A lumbar puncture should be performed if not contraindicated, and CSF should be assessed by flow cytometry, cytology, and immunoglobulin heavy-chain gene rearrangement. Because extraneural disease must be excluded to establish a diagnosis of PCNSL, computed tomography (CT) or CT/positron emission tomography (PET) scans of the chest, abdomen, and pelvis and a bone marrow biopsy and aspirate should be performed to exclude occult systemic disease. Involvement of the optic nerve, retina, or vitreous humor should be excluded with a comprehensive eye evaluation by an ophthalmologist that includes a slit lamp examination. Blood tests should include a complete blood count, a basic metabolic panel, serum lactate dehydrogenase, and HIV serology.
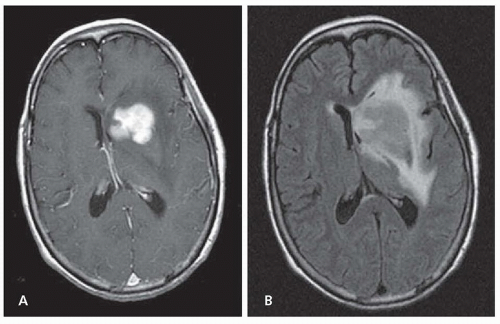 FIGURE 99.1 Magnetic resonance images from a patient with PCNSL. A T1-weighted, axial, postcontrast scan (A) demonstrates intense, homogenous enhancement of the tumor in the region of the left caudate nucleus. An axial T2/fluid-attenuated inversion recovery (FLAIR) scan at the same anatomic level (B) demonstrates hyperintense signal surrounding the tumor, reflecting vasogenic cerebral edema. (Courtesy of Priscilla K. Brastianos, MD.) |
Two prognostic scoring systems have been developed specifically for PCNSL. In a retrospective review of 105 PCNSL patients, the International Extranodal Lymphoma Study Group (IELSG) identified age older than 60 years, Eastern Cooperative Oncology Group (ECOG) performance status greater than 1, elevated serum lactate dehydrogenase (LDH) level, elevated CSF protein concentration, and involvement of deep regions of the brain as independent predictors of poor prognosis. In patients with zero or one factor, two or three factors, and four or five factors, the 2-year survival proportions were 80%, 48%, and 15%, respectively. In another prognostic model, PCNSL patients were divided into three groups based on age and performance status: (1) younger than 50 years old, (2) 50 years or older with a Karnofsky performance status (KPS) greater than or equal to 70, and (3) 50 years or older with a KPS less than 70. Based on these three divisions, significant differences in overall and failure-free survival were observed. There is no staging system that correlates with prognosis or response to treatment in PCNSL. However, because PCNSL is a multicompartmental disease potentially involving the brain, spinal cord, eye, and CSF, the IPCG recommends an extent of disease evaluation, as noted earlier, that will enable clinicians to follow the response to therapy.
TREATMENT
NEWLY DIAGNOSED PRIMARY CENTRAL NERVOUS SYSTEM LYMPHOMA
Treatment for newly diagnosed PCNSL consists of remissioninduction (induction) and remission-consolidation (consolidation) phases. Typically, induction consists of chemotherapy with the objective of achieving a complete response/remission. Once this response/remission is achieved, a different chemotherapy regimen or whole brain radiation therapy (WBRT) is administered to “consolidate” the response/remission. Defining response to treatment in PCNSL requires assessment of all sites (brain, CSF, eye) potentially involved by disease. The IPCG has established response criteria that have been adopted into most prospective clinical trials of PCNSL (Table 99.1).
Corticosteroids decrease tumor-associated edema and may result in partial radiographic regression of PCNSL. An initial response to corticosteroids is associated with a favorable outcome in PCNSL. However, after an initial response to corticosteroids, almost all patients quickly relapse. Corticosteroids should be avoided if possible prior to a biopsy, given the risk of disrupting cellular morphology, resulting in a nondiagnostic pathologic specimen.
Surgical resection is not part of the standard treatment approach for PCNSL given the multifocal nature of this tumor and has not been shown to prolong survival because most patients also receive additional treatment. Accordingly, the role of neurosurgery in PCNSL is to establish a diagnosis via stereotactic biopsy. However, it can be considered as a lifesaving intervention in extreme cases with large lesions and impending herniation.
Standardized induction and consolidation treatment for PCNSL has yet to be defined. Historically, PCNSL was treated only with WBRT at doses ranging from 36 to 45 Gy, which resulted in a high proportion of radiographic responses but also rapid relapse. In a multicenter phase 2 trial, 41 patients were treated with WBRT to 40 Gy plus a 20 Gy tumor boost and achieved a median overall survival (OS) of only 12 months. Given the lack of durable responses to radiation and the risk of neurotoxicity associated with this modality of therapy, WBRT alone is no longer a recommended treatment for most patients with PCNSL. Moreover, because PCNSL is an infiltrative, multifocal disease, focal radiation or
radiosurgery is not recommended. The most effective treatment for PCNSL is intravenous, high-dose methotrexate (HD-MTX) at variable doses (1 to 8 g/m2), typically used in combination with other chemotherapeutic agents and/or WBRT. However, there is no consensus on the optimal dose of HD-MTX or on the role of radiation in combination with methotrexate in the management of PCNSL. A number of randomized trials are ongoing to address these issues. Doses of methotrexate 3 g/m2 or higher result in therapeutic concentrations in the brain parenchyma and CSF and when combined with WBRT lead to more durable treatment responses. In a phase 2 trial, 79 PCNSL patients were randomized to receive either (1) HD-MTX (3.5 g/m2, day 1) versus (2) HD-MTX (3.5 g/m2, day 1) + cytarabine (2 g/m2 b.i.d., days 2 and 3) [Level 1].1 Each chemotherapy cycle was 21 days. All patients underwent consolidative WBRT after induction chemotherapy. The HD-MTX + cytarabine arm had a higher proportion of complete radiographic responses and a superior 3-year OS. However, it is now widely recognized that there is a high incidence of neurotoxicity with combined modality treatment that includes WBRT. The latter observation prompted studies using lower doses of WBRT. In a multicenter phase 2 study, no significant neurocognitive decline was observed after consolidative reduced-dose WBRT (23.4 Gy) and cytarabine in patients who had achieved a complete response to induction chemotherapy including HD-MTX. However, further study and longer neuropsychological follow-up of these patients is necessary to definitively assess the safety of this regimen because numerous studies have demonstrated the delayed neurotoxic effects of WBRT in the PCNSL population and the reduced risk of neurotoxicity in regimens consisting of chemotherapy alone. Given the risk of clinical neurotoxicity, other studies have assessed whether WBRT can be eliminated from the initial management of PCNSL. In a multicenter phase 3 trial, patients were randomized to receive HD-MTX-based chemotherapy with or without WBRT [Level 1].2 Five hundred fifty-one patients were enrolled, of whom 318 were treated per protocol. Intent to treat analysis revealed that patients treated in the combined modality arm (chemotherapy + WBRT) achieved prolonged progression-free survival (PFS) but no improvement in OS, demonstrating that the elimination of WBRT from the treatment regimen did not compromise OS. This has led to deferral of WBRT and the application of chemotherapy alone approaches for newly diagnosed PCNSL patients. These approaches are based on a foundation of HD-MTX. Variable doses and schedules of HD-MTX have been used, but in general, dose 3 g/m2 or higher delivered as an initial bolus followed by an infusion over 3 hours administered every 10 to 21 days is recommended for optimal outcomes and adequate CSF concentrations. Multiple phase 2 studies have demonstrated the safety, efficacy, and relatively preserved cognition of HD-MTX-based chemotherapy regimens. Moreover, longer duration of induction chemotherapy with HD-MTX (> 6 cycles) results in higher complete response proportions.
radiosurgery is not recommended. The most effective treatment for PCNSL is intravenous, high-dose methotrexate (HD-MTX) at variable doses (1 to 8 g/m2), typically used in combination with other chemotherapeutic agents and/or WBRT. However, there is no consensus on the optimal dose of HD-MTX or on the role of radiation in combination with methotrexate in the management of PCNSL. A number of randomized trials are ongoing to address these issues. Doses of methotrexate 3 g/m2 or higher result in therapeutic concentrations in the brain parenchyma and CSF and when combined with WBRT lead to more durable treatment responses. In a phase 2 trial, 79 PCNSL patients were randomized to receive either (1) HD-MTX (3.5 g/m2, day 1) versus (2) HD-MTX (3.5 g/m2, day 1) + cytarabine (2 g/m2 b.i.d., days 2 and 3) [Level 1].1 Each chemotherapy cycle was 21 days. All patients underwent consolidative WBRT after induction chemotherapy. The HD-MTX + cytarabine arm had a higher proportion of complete radiographic responses and a superior 3-year OS. However, it is now widely recognized that there is a high incidence of neurotoxicity with combined modality treatment that includes WBRT. The latter observation prompted studies using lower doses of WBRT. In a multicenter phase 2 study, no significant neurocognitive decline was observed after consolidative reduced-dose WBRT (23.4 Gy) and cytarabine in patients who had achieved a complete response to induction chemotherapy including HD-MTX. However, further study and longer neuropsychological follow-up of these patients is necessary to definitively assess the safety of this regimen because numerous studies have demonstrated the delayed neurotoxic effects of WBRT in the PCNSL population and the reduced risk of neurotoxicity in regimens consisting of chemotherapy alone. Given the risk of clinical neurotoxicity, other studies have assessed whether WBRT can be eliminated from the initial management of PCNSL. In a multicenter phase 3 trial, patients were randomized to receive HD-MTX-based chemotherapy with or without WBRT [Level 1].2 Five hundred fifty-one patients were enrolled, of whom 318 were treated per protocol. Intent to treat analysis revealed that patients treated in the combined modality arm (chemotherapy + WBRT) achieved prolonged progression-free survival (PFS) but no improvement in OS, demonstrating that the elimination of WBRT from the treatment regimen did not compromise OS. This has led to deferral of WBRT and the application of chemotherapy alone approaches for newly diagnosed PCNSL patients. These approaches are based on a foundation of HD-MTX. Variable doses and schedules of HD-MTX have been used, but in general, dose 3 g/m2 or higher delivered as an initial bolus followed by an infusion over 3 hours administered every 10 to 21 days is recommended for optimal outcomes and adequate CSF concentrations. Multiple phase 2 studies have demonstrated the safety, efficacy, and relatively preserved cognition of HD-MTX-based chemotherapy regimens. Moreover, longer duration of induction chemotherapy with HD-MTX (> 6 cycles) results in higher complete response proportions.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


