Disorder and OMIM link
Pathophysiology and inheritance
Typical features
Treatment
Association with spinal deformity
Rickets like disorders
Vitamin D-deficient rickets
Vitamin D deficiency from poor dietary intact, often breastfed, dark-skinned
Widened physes, lethargy, limb deformities
Physiological doses of vitamin D and calcium
Vitamin D-dependent rickets
#264700 (Type I)
#277440 (Type II)
%600785 (Type II with normal vitamin D receptor)
#193100 (autosomal dominant type)
Type I – AR, 12q13; genetic deficiency of enzyme 1-alpha-hydroxylase
Type II – abnormality of calcitriol receptor; end-organ resistance
Type I – low levels 1,25(OH)2 vitamin D
Type II – increased levels 1,25(OH)2 vitamin D; severe hypocalcemia
Type I – Calcitriol
Type II – Calcitriol, calcium
Spinal deformity rarely associated, possibly because of decreased growth velocities
X-linked hypophosphatemic rickets
#307800
Autosomal dominant
#193100
X-linked dominant, PHEX gene, Xp22 coding for a protease
AD, FGF23 gene, 12p13
Hypophosphatemia, decreased levels 1,25(OH)2 vitamin D;
normal calcium
Calcitriol; phosphate
Spinal deformity rarely associated, possibly because of decreased growth velocities
Lowe’s syndrome
#309000
Mutation in OCRL1 gene; Xp26; deficiency of phosphatidylinostitol 4,5-biphosphanate 5-phosphatase
Phosphaturia, hypophosphatemic rickets; aminoaciduria; decreased ammonia production by kidney – acidosis; carnitine wasting; cataracts; MR
Replacement therapy – phosphate, carnitine, alkali; supportive care
May develop increased lordosis and occasionally scoliosis [14]
Hypophosphatasia
#241500 (infantile)
#241510 (childhood)
#146300 (adult and odonthypophosphatasia forms)
Defect in alkaline phosphatase production
Identified in perinatal, infantile, childhood, adult, and odontohypophosphatasia forms. Perinatal hypophosphatasia lethal. Infantile form has a roughly 50 % mortality rate with symptoms appearing within the first 6 months after birth. The other forms are generally nonlethal. Adult form and odontohypophosphatasial form are marked by premature teeth loss. Common symptoms include bone malformations and higher chance of bone fracture
Osteogenesis imperfecta
Over 100 different mutations lead to OI
Incidence 1:10–20,000
Type I OI
166240 (IA), #166200 (IB)
AD. COL1A1 (chromosome 17) or COL1A2 (chromosome 7) functional null alleles causing reduced amounts of normal collagen I
Mildest and most common form of OI. Blue sclera, conductive hearing loss, with or without dentigenesis imperfecta
Bisphosphonate treatment currently with either pamindronate or alendronate for more severe cases
Occurrence of scoliosis is related to the severity of bone involvement
Type II OI
166210
Typically new mutation in either the COL1A1 gene or the COL1A2 gene
Fatal perinatally with intrauterine fractures, intracranial hemorrhage
Fatal too early for scoliosis
Type III OI
#259420
AD, rarely AR. Mutation in most cases lies in one of the genes for type I collagen, COL1A1 or COL1A2
One-eighth as common as type I. Severe involvement with progressive deformities, dentigenesis imperfecta, hearing loss, easy bruising, triangular facies
Bisphosphonate treatment currently with either pamindronate or alendronate
Scoliosis is very common. Kyphosis may also occur but is less common
Type IV OI
#166220
AD. COL1A1 or COL1A2
Similar to type I without blue sclera and more severe osseous involvement
Bisphosphonate treatment currently with either pamindronate or alendronate
Similar to type III
Type V OI
%610967
AD. Rare. Uncertain molecular mechanism
Bisphosphonate treatment currently with either pamindronate or alendronate
Similar to type III
Type VI OI
%610968
AD with parental mosaicism. Rare. Uncertain molecular mechanism
[17] Similar to type IV
Bisphosphonate treatment currently with either pamindronate or alendronate
Similar to type III
Type VII OI
#610682
Eight affected individuals in a small consanguineous First Nations community in northern Quebec. Mutation in the CRTAP gene (also cause type IIB)
Bone fragility and low bone mass
Uncertain
Uncertain
Type VIII OI
#610915
AR. Mutation in the gene encoding leprecan (LEPRE1)
Lethal
Uncertain
Uncertain.
Type IX OI
#259440
AR. Abnormality in PPIB gene
Lethal
Uncertain
Uncertain
Type X OI
#613848
AR. Mutation in the serpin peptidase inhibitor (SERPINH1)
Lethal
Uncertain
Uncertain
Type XI OI
#610968
AR. Homozygous mutation in the FKBP10 gene
Severe OI without dentogenesis imperfecta
Uncertain
Scoliosis in 5/8 reported patients
Type XII OI
#613849
AR. Mutation in the SP7 gene
Milder deformity without dentogenesis imperfecta. White sclera
Uncertain
Scoliosis reported
Type XIII OI
#614856
AR. Homozygous mutation in the BMP1 gene
Variable
Uncertain
Kyphoscoliosis (in some patients). S-curve scoliosis of thoracic and lumbar spine (in some patients)
Type XIV OI
#615066
AR. Homozygous mutation in the TMEM38B gene
Variable
Uncertain
Uncertain
Type XV OI
#615220
AR. Homozygous or compound heterozygous mutation in the WNT1 gene
More severe, blue sclera
Uncertain
Scoliosis reported
Other osteopenic syndromes
Bruck syndrome #259450
Rare. Possibly gene encoding bone telopeptidelysyl hydroxylase
Uncertain
Severe scoliosis similar to OI type III
Osteoporosis-pseudoglioma syndrome #259770
Familial gene encoding low-density lipoprotein receptor-related protein-5
Blindness. Brittle bones
Uncertain
Scoliosis is reported
Idiopathic juvenile osteoporosis #259750
Uncertain etiology
Idiopathic osteoporosis which resolves with adolescence
Uncertain. Protect spine
May develop spine compression fractures
Selected Mucopolysaccharidoses | ||||
Mucopolysaccharidoses | Pathophysiology | Typical features | Treatment | Association with spinal deformity |
Type I (Hurler’s syndrome) #607014 | Gene encoding alpha-l-iduronidase | Course features are evident early. Bulging fontanels, neurological compression, corneal clouding; upper airway obstruction; pulmonary edema postoperative; short stature; carpal tunnel | Bone marrow transplant Enzyme therapy may help ? Gene therapy | Gibbus deformity lower spine. Upper cervical instability, odontoid hypoplasia and compression from instability or dural and ligamentous hypertrophy may occur |
Type II (Hunter syndrome) +309900 | Deficient activity of iduronate 2-sulfatase X-linked; mapped to Xq27–28 Dx: Enzyme assays in cultured fibroblasts/leukocytes Increased urinary heparan and dermatan sulfate | Two forms exist (Type A – severe; Type B – mild) Type A – clinical features as in Type IH; onset age 1–2 years; death in adolescence, third decade Type B – may be diagnosed in adulthood Course facial features; hearing loss; mental retardation (Type A); absence of corneal clouding; upper airway obstruction; pulmonary edema postoperative; HSM; dysostosis multiplex; short stature; HCP; carpal tunnel; ivory skin lesions; Mongolian spots; hypertrichosis | Bone marrow transplant Enzyme therapy may alter disease progression; not curative Idursulfase ? Gene therapy | Similar to other mucopolysaccharidosis (MPS) |
Type III (Sanfilippo syndrome) #252900 #252920 #252930 #252940 | Type A – deficiency of heparin N-sulfatase (17q25.3) Type B – deficiency of alpha-N-acetylglucosaminidase (17q21) Type C – deficiency of aceyl-CoA:alpha-glucosaminide acetyltransferase (14) Type D – deficiency of N-acetylglucosamine-6-sulfatase (12q14) Dx: Increased urinary heparan sulfate | Type A – most severe, aggressive Behavioral issues at 2 years, neurological manifestations at 6 years Death in second decade Corneal clouding not a common finding; attentional/behavioral problems; hyperactivity; seizures; diarrhea; URI | Supportive care Not improved by bone marrow transplant | Similar to other MPS |
Type IV (Morquio syndrome) Numerous types | Type A – deficiency of galactosamine-6-sulfatase (GALNS gene, 16q24.3) Type B – β-galactosidase (GLB1 gene, 3p21.33) Increased urinary excretion of keratin sulfate (cartilage/cornea) mild may not excrete ELISA Enzyme assay in cultured fibroblasts/leukocytes Genetic testing to detect mutations in GALNS, GLB1 | Clinically, both forms may be similar; great variability in severity within both groups Mortality related to atlantoaxial instability, myelopathy and pulmonary compromise For severe, death in second or third decade of life No course facial features; normal intelligence; spondyloepiphyseal dysplasia; ligamentous laxity; odontoid hypoplasia; shortened trunk dwarfism; genu valgus; greater incidence of spinal involvement; bowel/bladder incontinence; OSA; pulmonary infections – chest wall deformity; heart valve thickening/defects; corneal clouding; enamel Less common – hearing loss, hernias | Supportive treatment | Similar to other MPS. These patients survive long enough that orthopedic treatment, particularly for cervical instability may be necessary |
Type VI (Maroteaux–Lamysynd) #253200 | AR Deficiency of N-acetylgalactosamine 4-sulfatase Accumulation of dermatan sulfate | Acceleration in growth during first year; followed by regression and short stature; course features, HSM, corneal clouding; normal intelligence Other: hearing loss, respiratory infections, valvular disease | Bone marrow transplant reported as successful. Enzymatic treatment approved | Similar to other MPS |
Type VII (Sly disease) #253220 | Deficiency of β-glucoronidase, required for degradation of dermatan sulfate, heparin sulfate, and chondroitin sulfate Mutation in gene mapped to chromosome 7 | Phenotype varies; severe form (subtype 1) present at birth – jaundice, anemia, hydrops; milder forms present later, before (subtype 2) or after (subtype 3) age 4 years; course features; HSM; hernias; MR; dysostosis multiplex | Bone marrow transplant reported as successful in improving daily function but not the mental retardation | Similar to other MPS |
Pulmonary failure is the leading cause of death in adults with OI, and it is closely associated with thoracic scoliosis. Widmann et al. [19] found a high negative correlation between pulmonary function and thoracic scoliosis and but not with chest wall deformity or kyphosis. They also found diminution in vital capacity below 50% in thoracic curve of 60° or more.
Spinal deformity in OI is directly related to the severity of osseous involvement [20–22], though body weight is also a factor in bone density [23]. Anissipour et al. [20] recently evaluated progression of scoliosis in a large cohort of patients with various types of OI and found the most rapid progression in the more severe type III. While ligamentous laxity, also from type I collagen deficiency, may also contribute, a series of patients with various types of OI found less scoliosis in those with ligamentous laxity [21]. There is a tendency for later development of scoliosis in those with early development of motor milestones, particularly supported sitting [21]. Overall, the worse the osseous involvement in terms of intrinsic vertebral body deformity and decreased Z-scores for bone density, the worse the scoliosis and the more difficultly achieving good fixation for correction [22, 23]. The morphology of the vertebral bodies ranges from normal contours to flattened, wedged, and biconcave with the biconcave vertebra more likely to develop severe scoliosis [23–25]. Six or more biconcave vertebra before puberty appears prognostic of developing scoliosis of greater than 50° [23]. The spine may also become kyphotic particularly [26] in those with more severe involvement [27]. Cervical fractures [28] including spontaneous paraplegia from chiropractic manipulation [29], thoracic fractures with spinal cord injury [30], and multiple flexion stress endplate fractures of the thoracic and lumbar spine [31] have been reported. Daivajna et al. [32] reported a modified anterolateral approach for decompression of myelopathy from severe cervical kyphosis in a 9-year-old with OI.
Bracing has been attempted for spinal deformity from OI. However, the corrective force applied through the pathological ribs only leads to further rib deformity and may contribute to worsening pulmonary function [33–35]. Current braces cannot achieve this. Patients who are not operative candidates can be fitted with custom seating for comfort and function [36, 37]. Bracing’s role in OI is primarily limited to postoperative temporary support though there is some suggestion that an orthosis can slow the progression of basilar impression [3].
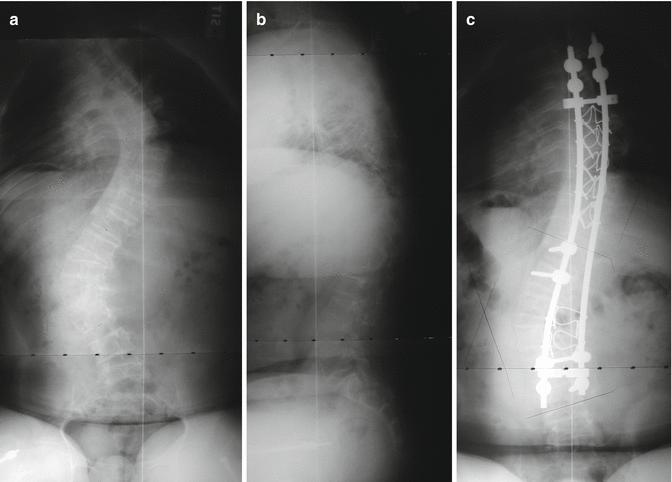
Positioning patients for surgery can be very difficult and must be carefully supervised because of chest wall deformities, fragile extremities, ribs, and frequent contractures. Unpadded blood pressure cuffs can result in fractures. Surgery for these patients should typically occur in institutions accustomed to their care [38]. Fatality has been reported from intraoperative rib fractures [39]. Classically, correcting curves in severely soft bone has been difficult at best with most authors recommending stabilization rather than relying upon correction [34]. The stability of fixation depends upon both the strength of the bone and the quality of the fixation’s purchase. Polymethylmethacrylate has been used for additional fixation [33, 40, 41]. Historically, each new type of fixation has been attempted. As might be expected, the greater the purchase and the more segmental the fixation, the less stress required on any single level and the greater the correction achieved [42] (Figs. 19.1a–c and 19.2a–d). Preoperative halo gravity traction using multiple pins may be useful [43, 44] though both sixth and fourth nerve palsies have been reported in OI patients [45]. Postoperatively, there is little change from preoperative ambulatory ability or activity [34, 37, 46], though patients report less pain, fatigue, and dyspnea [47], and the improvements gained at surgery are usually maintained into adulthood [43, 47]. Spondylolysis [48] or an elongated pars with spondylolysthesis [49] can occur below a long spinal fusion. Anterior interbody fusion has been described [49], but the spondylolisthesis may not be symptomatic (Fig. 19.3).
 Other Neuromuscular Diseases
Other Neuromuscular Diseases
 Clinical Examination and Associated Comorbidities of Early Onset Scoliosis
Clinical Examination and Associated Comorbidities of Early Onset Scoliosis
 Revision Spine Surgery in the Growing Child
Revision Spine Surgery in the Growing Child
 Convex Growth Arrest for Congenital Scoliosis
Convex Growth Arrest for Congenital Scoliosis
 Growth-Guided Instrumentation: Shilla Procedure
Growth-Guided Instrumentation: Shilla Procedure
 Plate-Rod System in the Management for Progressive Scoliosis in Growing Children
Plate-Rod System in the Management for Progressive Scoliosis in Growing Children




Related posts:
Other Neuromuscular Diseases
Clinical Examination and Associated Comorbidities of Early Onset Scoliosis
Revision Spine Surgery in the Growing Child
Convex Growth Arrest for Congenital Scoliosis
Growth-Guided Instrumentation: Shilla Procedure
Plate-Rod System in the Management for Progressive Scoliosis in Growing Children
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
