and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
18.1 General Aspects of Toxic Neuropathy
Exogenous substances that can cause peripheral nerve injury include pharmaceutical agents, heavy metals, industrial chemicals, and nutritional and biological toxins. If a history of toxic exposure is not obtained or an alternative etiology is under consideration, nerve biopsy might be performed.
A discussion of each of the many substances that are toxic to the peripheral nerve is beyond the scope of this book (see recent review, Manji 2013; Diezi et al. 2013). As most human toxic neuropathies show nonspecific histological changes, we will concentrate on a small number of neuropathies that have unusual pathological features or have been extensively studied and serve as paradigms for toxic nerve injury. Tables 18.1 and 18.2 provide a broader listing.
Table 18.1
Toxic neuropathies caused by pharmaceutical agents
Toxin | Used for | Clinical features | Pathological features and comments |
|---|---|---|---|
Almitrine | COPD | Sensory, chronic | |
*Allopurinol | Gout Rx | Sensory > motor, chronic | |
Amiodarone | (See text) | ||
*Amitriptyline | Antidepressant, suicide attempts | Acute severe sensorimotor neuropathy | |
Bortezomib | Chemotherapy | Acute severe mostly sensory small fiber predilection | |
*Chloramphenicol | Antibiotic | Sensory + optic | No human material (Joy et al. 1960) |
Chloroquine | Antimalarial | Myopathy > neuropathy | |
Cisplatin | Chemotherapy | Sensory > motor | |
Colchicine | Gout, FMF, Behçet’s syndrome | Myopathy > neuropathy | Axonal, involves large MFs > small MFs with evidence of axonal degeneration, regeneration (Kuncl et al. 1987) |
Sensory > motor | |||
Dapsone | Leprosy, malaria, rheumatoid arthritis, pneumocystis pneumonia, dermatologic | Motor > sensory | Axonal degeneration (Navarro et al. 1989) |
ddI/ddC | HIV Rx | Distal sensory > motor | Electrophysiology indicates an axonal process (Berger et al. 1993) |
Disulfiram | (See text) | ||
Doxorubicin | Antineoplastic | Not described in humans | Sensory neuronopathy through spinal ganglion injury (animal data) (Cho 1977) |
Ethambutol | Antituberculous | Sensory > motor | |
*FK506 | Immune suppressant | CIDP-like picture | Axonal depletion, demyelination, no inflammation (1 report only) (Wilson et al. 1994) |
Gold | Rheumatoid arthritis | Subacute motor, sensorimotor neuropathy, myokymia | Axonal > demyelinating (Katrak et al. 1980) |
*Hydralazine | Antihypertensive | Sensorimotor | No biopsy data. Possible role of hydralazine-induced pyridoxine deficiency (Raskin and Fishman 1965) |
Isoniazid | Antituberculous | Sensory > motor | |
*Lithium | Mood stabilizer | Acute–subacute sensorimotor | Axonal. Occurs most convincingly after acute intoxication (Vanhooren et al. 1990) |
L-tryptophan | (See text) | ||
Metronidazole | Antibiotic, antiprotozoal, radiation sensitizers | Sensory > motor, large fiber | Axonal degeneration and demyelinating, relative sparing of UFs (Takeuchi et al. 1988) |
Misonidazole | (See text) | ||
Nitrofurantoin | Antibiotic (urinary) | Subacute sensorimotor, potentially severe. Higher risk in renal failure | Axonal, involving large MFs most affected (Yiannikas et al. 1981) |
Nitrous oxide | Mild anesthetic, abuse | Sensory > motor polyneuropathy and myelopathy (SCD-like) | |
Perhexiline | Antianginal | Sensorimotor | |
APhenytoin | Anticonvulsant | Sensorimotor, usually subclinical | Axonal, large MFs most affected, ?secondary demyelination (Ramirez et al. 1986) |
Cis-platinum | Antineoplastic | Large > small fiber sensory | |
Podophyllin | Herbal remedy, topical agent | Severe acute–subacute sensorimotor | |
*Procarbazine | Antineoplastic | Mild–moderate sensorimotor | Not available (Samuels et al. 1967) |
Pyridoxine | Vitamin abuse | Sensory, large > small fiber | Axon loss/atrophy, DRG neuron loss, ? blockade of fast axonal transport in the proximal axon (Schaumburg et al. 1983) |
Sodium cyanate | Sickle cell anemia | Sensorimotor | |
Suramin | Antineoplastic and antiparasitic | Severe sensorimotor | Mixed axonal and demyelinating; known inhibitor of lysosomal enzymes which accumulates in DRG (LaRocca et al. 1990; Chaudhry et al. 1996). The mechanism is unestablished, but suramin is a known inhibitor of lysosomal enzymes and accumulates in DRG neurons in experimental animals; in addition, suramin blocks the receptor function of IGF-I and NGF |
Tacrolimus | Immune suppression | Sensorimotor | Axonal loss and demyelination, CIDP mimic |
Taxanes (paclitaxel, docetaxel) | Antineoplastic | Distal sensory > motor axonopathy | MF and UF loss, axonal atrophy, membranotubular aggregates but without microtubule aggregates (occur in cultured cells, localized nerve injection) and with inconstant evidence of regeneration. Calpain inhibitors may inhibit axonal degeneration in experimental animals. NGF, IGF-I, ORG 2766, and LIF are reported to protect against the neuropathy (Lipton et al. 1989; Sahenk et al. 1994; Herskovitz and Schaumburg 2005) |
Thalidomide | Behçet’s disease, graft vs. host Dz, skin disease, leprosy, HIV, plexiform neurofibromas, Crohn’s disease, and as an antiangiogenic antitumor, immune-modulating agent | Sensory > motor | Axonal degeneration, large MFs most affected (Chapon et al. 1985; Fullerton and O’Sullivan 1968; Chaudhry et al. 2002; Fleming et al. 2005; Giannini et al. 2003). Painful paresthesias of the hands and feet. Axonal degeneration and loss of large MFs, regenerating axons, increased numbers of regenerating axons, and loss of dorsal root ganglia neurons, a pattern consistent with dying-back neuropathy and/or with sensory ganglionitis |
Thiophenicol | Antibiotic | Subacute, sensory | ? demyelinating (Shinohara et al. 1977) |
Vincristine | (See text) |
Table 18.2
Neuropathy caused by metals and biological/occupational/household toxins
Toxin | Source | Clinical features | Pathological features |
|---|---|---|---|
Acrylamide | Grouting/strengthening agent in mining and paper industries | Chronic sensorimotor | Human data suggests distal axonopathy with giant axonal filamentous changes, although not to same extent as hexacarbon toxicity. May reflect interference with slow and fast axonal transport; inhibiting fast motor kinesin interaction with microtubules. Experimental data is similar (Davenport et al. 1976; Fullerton 1969; Gold et al. 1985; Schaumburg and Berger 1993; Berger and Schaumburg 2005) |
Arsenic | Antimicrobial, poisoning | Acute sensorimotor | Axonal, with large MFs most involved, minimal segmental demyelination (Donofrio et al. 1987) |
Buckthorn | Shrub endemic to SW North America | Acute severe sensorimotor | |
Carbon disulfide | Rayon manufacture | Chronic sensorimotor | |
Cyanide | Cassava, sorghum, laetrile | Chronic distal sensorimotor | |
Dimethylaminopropionitrile | Grouting agent, polyurethane foam manufacturing | Sensorimotor, small fiber | Axonal, possibly involving UFs most prominently with particulate organelle hyperplasia and neurofilament disorganization (Pestronk et al. 1980) |
Diphtheria toxin | (See Sect. 11.5) | ||
Ethylene oxide | Cool sterilization, industrial chemical manufacture | Sensorimotor neuropathy; encephalopathy | |
Lead | (See text) | ||
Mercury | Industrial pollutant, accidental exposure | Sensorimotor neuropathy, encephalopathy | Axonal, probably a neuronopathy (Windebank 1993) |
Methyl methacrylate | Resin (dental, orthopedic) | Sensorimotor, chronic | Axonal, involving large MFs > small MFs (Donaghy et al. 1991) |
Methyl n-butyl ketone | (See text – Sect. 18.7) | ||
N-Hexane | (See text – Sect. 18.7) | ||
Organophosphate | Insecticides, plastics, petroleum additives | Acute/subacute sensorimotor | |
Thallium | Rat poison, depilating agent | Acute/subacute sensorimotor, often painful | |
Vacor | Rat poison | Hyperacute sensorimotor neuropathy |
Axonal degeneration underlies nearly all toxic neuropathies, and in many instances, a distal axonopathy has been identified. The histological features of axonal degeneration are discussed in detail in Chap. 4. Several principles are helpful in understanding the clinical and histological features of toxic neuropathies:
1.
Axons are affected earlier than Schwann cells, probably because the complexity of maintaining an axon that is as long as a meter from the cell body is greater than that of maintaining a myelinated internode. Exceptions to this rule arise when the toxin specifically upsets some aspect of myelin or Schwann cell metabolism, such as diphtheria toxin or the amphiphilic drugs amiodarone and perhexiline.
2.
Large myelinated axons are affected first and most severely. The larger the volume of axoplasm, the more challenging the metabolic task of maintaining the axon and the more vulnerable the axon is to exogenous toxins. Examples of possible exceptions to this rule are dimethylaminopropionitrile (Pestronk et al. 1980) and pyridoxine exposure (Berger et al. 1992). It is possible that this observation has been overemphasized because of difficulties in quantitating unmyelinated fiber injury pathologically and electrophysiologically.
3.
There is usually a correlation between the dose and duration of intoxicant exposure and rate of onset and severity of neuropathy. However, this is not always evident, perhaps due to interindividual differences in drug metabolism. Some neuropathies seem to occur only after massive exposure to the toxin (lithium, amitriptyline), while others occur only after many years of drug use (anticonvulsants).
In keeping with these tenets, most toxic neuropathies are length dependent and affect predominantly large axons. This has certain clinical consequences. The longest peripheral nerve axons in the body are sensory fibers to the toes, explaining why distal paresthesias often herald a neuropathy well before the appearance of weakness. Thus, unless permitted to progress despite early symptoms, most toxic neuropathies are predominantly sensory. Furthermore, alteration in large fiber sensation, i.e., vibration, is an early objective finding. Cranial nerves are almost never involved, but evidence of CNS dysfunction is sometimes present. The tempo of progression depends on the duration and dose of exposure, as well as interindividual variations in metabolism, and is usually not characteristic for a particular toxin. Nerve conductions confirm that a predominantly axonal process is occurring, and most ancillary tests, including CSF examination, are normal. There are, of course, exceptions to all these generalizations. Amphiphilic drugs (amiodarone, perhexiline) cause a demyelinating or mixed neuropathy, and thus motor involvement is early, and nerve conduction may be slowed. Secondary demyelination can result in prominent conduction slowing in severe hexacarbon neuropathy (vide infra). Gold, arsenic, and nitrofurantoin are particularly liable to cause an acute–subacute neuropathy.
Prognosis is usually good if the toxin is withdrawn, with axonal neuropathies recovering to a variable extent over months to years and demyelinating neuropathies more rapidly and completely.
18.2 Amiodarone
18.2.1 Clinical Manifestations
Initially used in the treatment of angina nearly three decades ago, amiodarone is presently employed with increasing frequency in the treatment of cardiac arrhythmia. Although not mentioned in one large review (Raeder et al. 1985), some studies indicate that 6–10 % of amiodarone-treated patients develop a peripheral neuropathy (Charness et al. 1984; Palakurthy et al. 1987). Usually, the neuropathy occurs in a patient who has been on the drug for 6 months or more, at doses of 200–600 mg/day. However, onset after 1 month with high doses (1,400 mg/day) and many months with very low doses (50 mg/day for 18 months) have been described (Pellissier et al. 1984). Progression is typically over weeks to months, with symmetrical distal paresthesias, followed by diminished motor and sensory function. Reflexes are diminished or absent. Myopathy often accompanies the neuropathy. CSF protein can be elevated, occasionally markedly (Pellissier et al. 1984). Conduction studies may show elements of both axonal loss and demyelination, but either can predominate, and denervation is commonly seen on EMG. If the clinician is not aware of the patient’s use of this medication, an incorrect diagnosis of Guillain–Barre syndrome or CIDP is possible. Most workers report improvement within 1–6 months of stopping the drug. This has also been our experience with three cases.
It is noteworthy that an examination of the incidence of polyneuropathy in patients treated with a variety of antiarrhythmic drugs suggested that this occurred no more frequently with amiodarone than with other antiarrhythmics and that clinically significant neuropathy was rare (Collaborative 1994). However, the average dose (200 mg) of amiodarone used was small.
18.2.2 Pathology
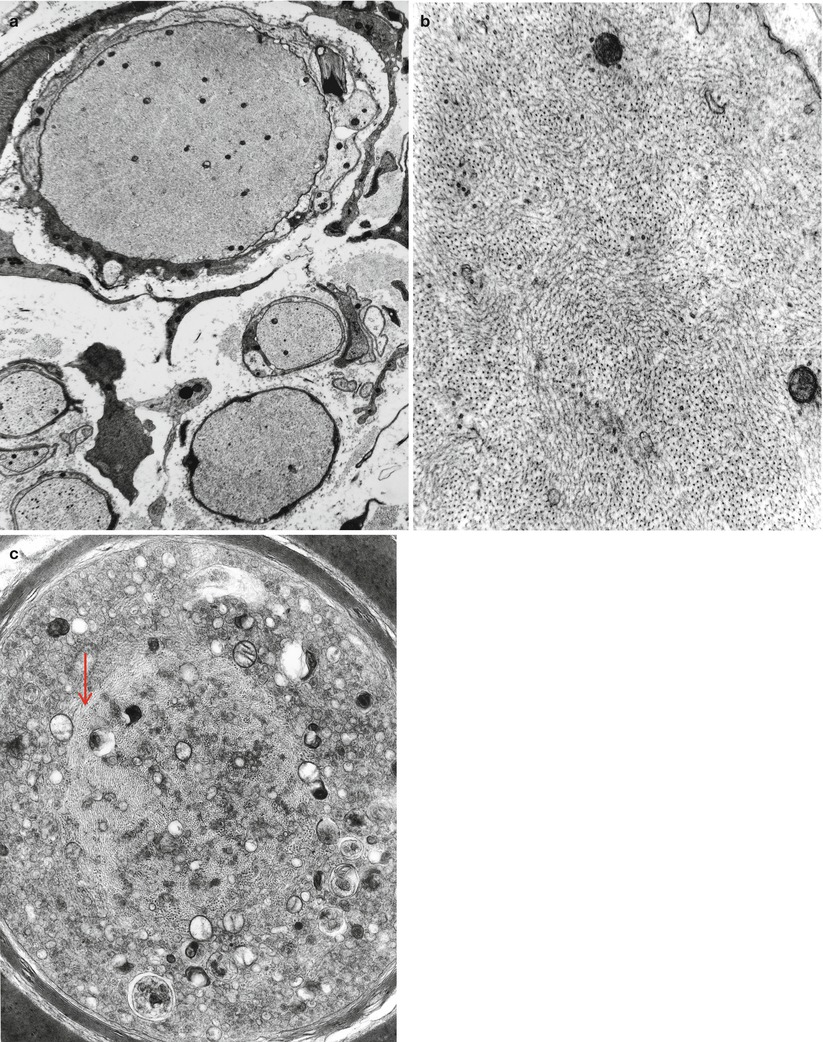
The characteristic peripheral nerve finding in amiodarone neuropathy is an accumulation of lysosomal inclusions. These are found in great numbers in endothelial cells, vascular smooth muscle cells, pericytes, perineurial cells, fibroblasts, and Schwann cells, especially nonmyelinating Schwann cells (Figs. 18.1, 18.2, 18.3, 18.4, and 18.5). We have rarely observed them within axons (Fig. 18.5). These inclusions are well visualized in semithin toluidine blue-stained sections (Figs. 18.1 and 18.2) but are not retained in paraffin-embedded material.
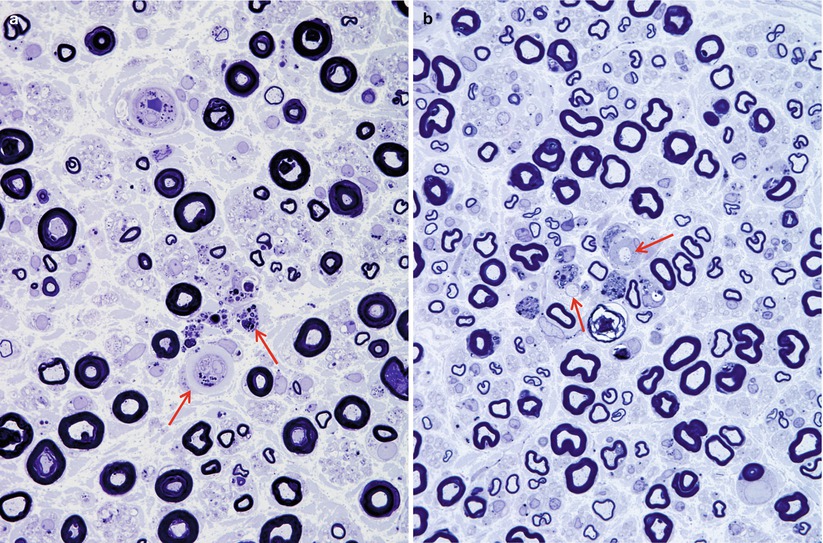
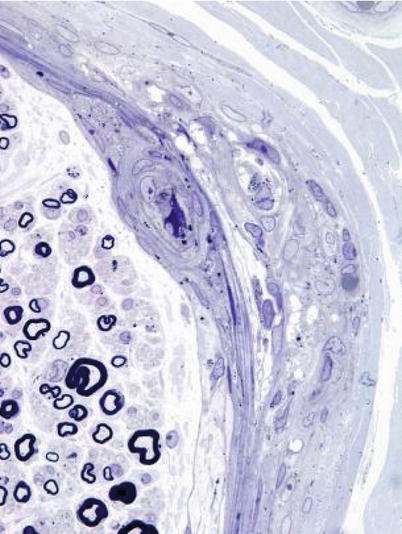
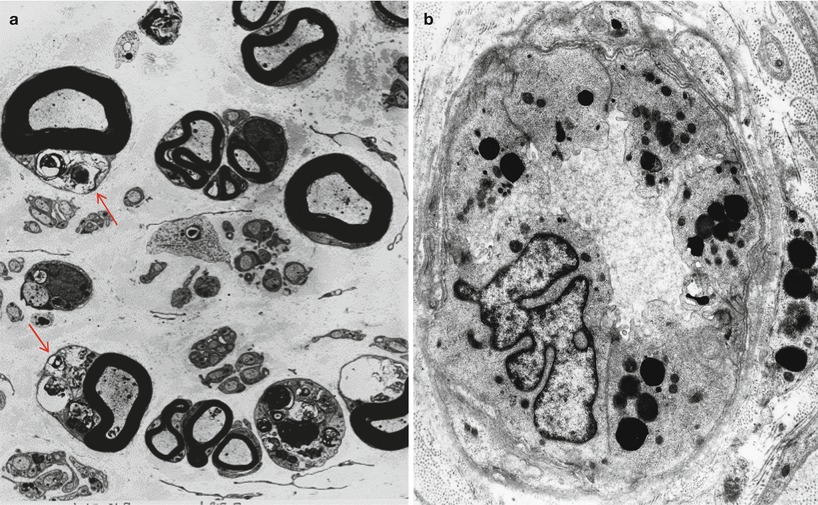
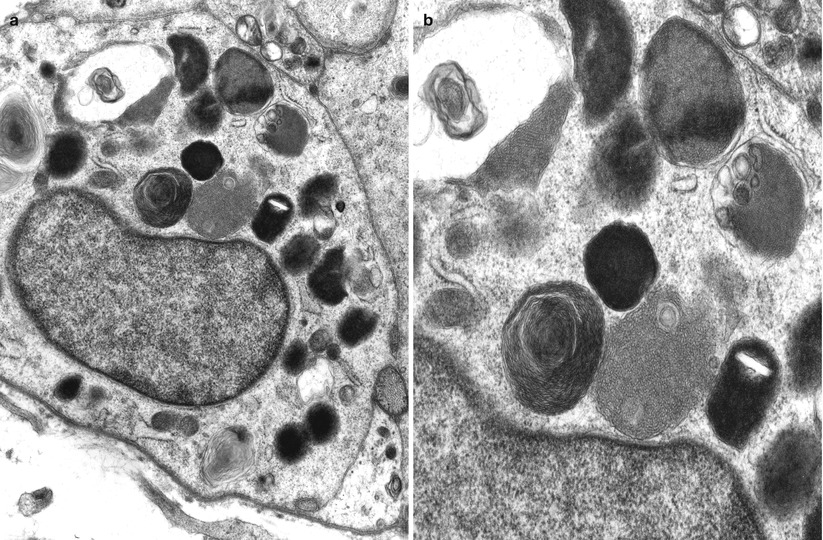
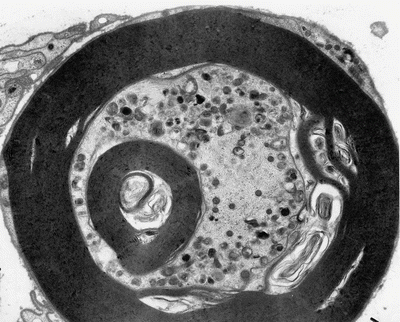
Fig. 18.1
Amiodarone: note loss of myelinated fibers and small lipid inclusions in vessels and endoneurial cells (arrows, a). Several denuded axons are shown (arrows, b) (1 μm thick plastic sections; magnification: a, b 1,000×)
Fig. 18.2
Amiodarone: focal damage to fascicle including perineurium is seen. Note scattered osmiophilic bodies in perineurium and blood vessels (1 μm thick plastic section; magnification 1,000×)
Fig. 18.3
Amiodarone: numerous cytoplasmic osmiophilic inclusions are seen in Schwann cells (arrows, a) of degenerating and demyelinated axons. (b) Inclusions are prominent in endothelium and pericytes (a, 3,000×; b, 14,910×)
Fig. 18.4
Amiodarone: lysosomal inclusions of varying morphologies are shown in Schwann cells of denervated band (a) and at higher magnification (b) (a, 14,560×; b, 25,000×)
Fig. 18.5
Amiodarone: note accumulation of lamellar and dense osmiophilic vesicles in axon, with appearance of axonal atrophy and myelin irregularities (12,070×)
The pathological process is usually active axonal degeneration (Fig. 18.1a and 18.3) with variable amounts of demyelination (Figs. 18.1b and 18.3a) (Pellissier et al. 1984). Axonal loss ranges from mild to subtotal and can affect large myelinated fibers preferentially, with relative sparing of unmyelinated fibers (Meier et al. 1979; Jacobs and Costa-Jussa 1985; Pellissier et al. 1984). Regenerating clusters may be readily seen (Fig. 18.3a). It is the occasionally prominently demyelinating nature of this neuropathy that sets it apart from most other toxic exposures. In our experience, the numbers of thinly myelinated fibers, debris-filled macrophages, large naked axons, and even onion-bulb formations have varied considerably between biopsies. Some authors have reported very prominent segmental demyelination with little or no axon loss (Jacobs and Costa-Jussa 1985). In one instance, we observed focal perineurial disruption with axonal degeneration in the adjacent endoneurium (Fig. 18.2).
Electron microscopy reveals that the cytoplasmic inclusions are 0.5–1.5 μm in size and consist of membrane-bound osmiophilic material (Figs. 18.3b and 18.4a, b). The inclusions may take a lamellar or granuloreticular/crystalline appearance. The former may demonstrate a circular or linear, tightly or loosely packed lamellar structure (Fig. 18.4b). In our experience, there is no association between cell and inclusion types, although some authors have claimed otherwise (Pellissier et al. 1984). Moreover, it is not difficult to find inclusions which show features of both ultrastructural patterns (Fig. 18.4b), suggesting that the two inclusion types are related, and the different morphology may reflect different stages of evolution.
Jacobs and Costa-Jussa (1985) observed increased amounts of a finely granular Schwann cell cytoplasm with diminished numbers of organelles, even in the absence of inclusions. It was reported that similar alterations were seen in endothelial and perineurial cells and postulated that these changes may precede the formation of inclusions. The axoplasm itself often shows nonspecific changes (filamentous hyperplasia, crystalloid osmiophilic filamentous aggregates, and glycogen deposits), although typical inclusions are uncommon (Fig. 18.5).
18.2.3 Pathogenesis
Amiodarone belongs to the class of amphiphilic substances, which include chloroquine (Figs. 18.6, 18.7a, b, and 18.8a, b) and perhexiline, and are well known to cause systemic formation of myelinoid bodies (lamellar, granuloreticular or crystalline) having all the characteristics discussed above for amiodarone (Hruban et al. 1972; Hruban 1984; Lullmann et al. 1978). The inclusions appear to reflect lysosomal accumulation of membranes whose degradation is impaired by the toxin (Hruban et al. 1972; Hruban 1984). Amiodarone accumulates within affected tissue (Meier et al. 1979; Fraser et al. 1985) and likely forms a drug–phospholipid complex which resists degradation; amiodarone appears to block the action of phospholipase A (Hruban 1984; Kannan et al. 1991).
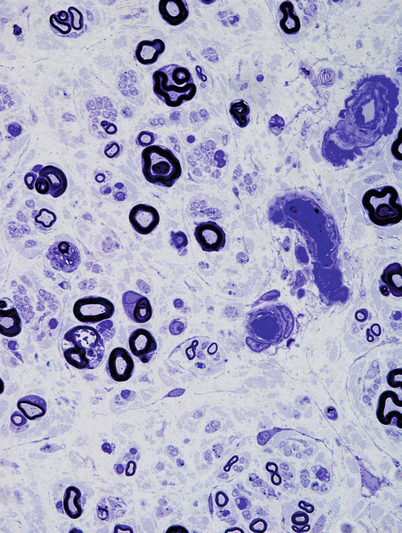
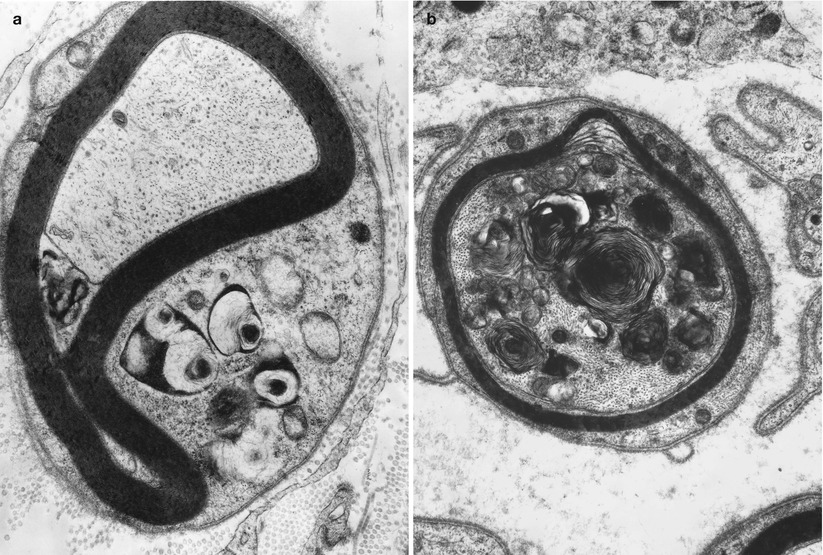
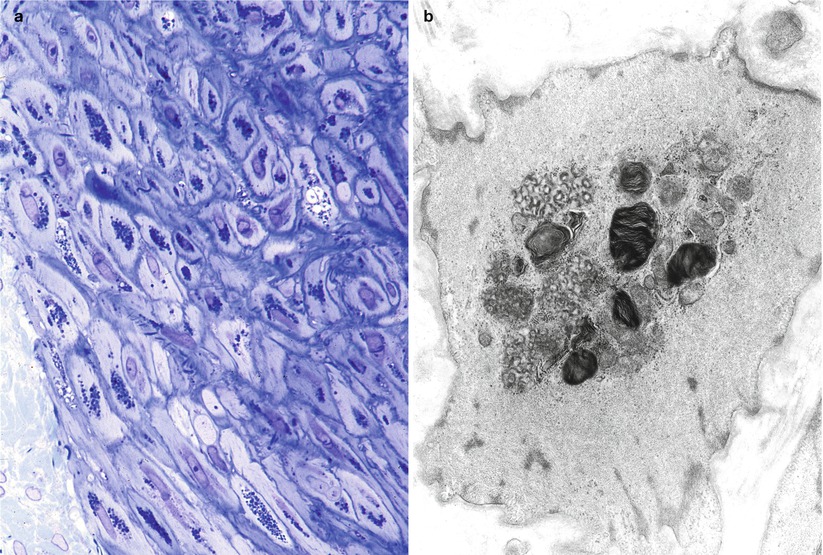
Fig. 18.6
Chloroquine neuropathy: characterized by significant axon loss and active axonal degeneration (1 μm thick plastic section, 1,000×)
Fig. 18.7
Chloroquine neuropathy: compact and loose osmiophilic lamellar inclusions in Schwann cell cytoplasm (a) and within myelinated axon (b) (Magnification: a, 16,800×; b, 22,880×)
Fig. 18.8
Chloroquine epineurial vessel shows accumulation of osmiophilic inclusions in smooth muscle cells (a, 1 μm thick plastic section, 400×; b, 15,000×)
The mechanism by which axons or Schwann cells are damaged remains unknown. Jacobs and Costa-Jussa (1985) postulated a toxic schwannopathy based on case material that showed dominant demyelination with early Schwann cell cytoplasmic changes. Most authors, however, have shown a significant axonal loss which remains unexplained.
18.2.4 Differential Diagnosis
The inclusions of amiodarone neuropathy, whether lamellated or paracrystalline, are nonspecific, simply reflecting a disturbance of lysosomal function. Several storage diseases should be considered.
Any of the causes of systemic phospholipidosis may demonstrate similar inclusions (Hruban 1984), but in the setting of neuropathy, perhexiline and chloroquine toxicity should be considered. If the history of exposure to amiodarone is missed, the clinician will usually be considering the diagnosis of inflammatory demyelinating neuropathy, and this issue is easily resolved once the characteristic inclusions are observed. Neuropathy in patients taking amiodarone has been described in the absence of inclusions (Dubois et al. 1984; Vital and Vallat 1987a, p185). It remains unclear whether or not these patients suffered from another cause of peripheral neuropathy (e.g., hypothyroidism due to amiodarone or an unrelated cause) or whether it is possible to have significant neuropathy without characteristic inclusions. In our experience, the inclusions are so abundant that we would be reluctant to consider the possibility of an amiodarone neuropathy in their absence.
Fabry and Niemann-Pick diseases also demonstrate intracellular storage in a variety of endoneurial cells and under light microscopy may be superficially similar to amiodarone neuropathy (see Chap. 20). One helpful distinguishing point is the rarity of Fabry inclusions in Schwann cells. The inclusions of Fabry disease demonstrate the “Maltese cross” pattern on frozen sections under polarized light and are larger than those of amiodarone even on light microscopy. Paracrystalline inclusions are not a significant feature of Fabry or Niemann-Pick disease but are abundant in amiodarone neuropathy. These are also seen in small numbers in a variety of neuropathies. Zebra bodies are not seen in amiodarone neuropathy. The osmiophilic storage material of Fabry disease usually contains regions with regular 4–6 nm periodicity. In Niemann-Pick disease, peripheral nerve inclusions may take several forms, most often amorphous and lipopigment-like. Other storage diseases including metachromatic leukodystrophy, globoid cell leukodystrophy, and adrenoleukodystrophy have very characteristic inclusion ultrastructure and will not pose a diagnostic problem (Chap. 20).
Case 18.1
Four weeks prior to presentation, a 50-year-old male noted numbness and tingling in his feet followed by progressive weakness in the lower extremities to the point where he became bed-bound. Mild weakness in the arms was also present. Medical history was notable for several myocardial infarctions, congestive failure, and cardiac arrhythmia. Medications included amiodarone, which had been started at 600 mg/day 3 months previously. Examination disclosed an alert man with normal cranial nerve function. There was diffuse grade 4+/5 (MRC) weakness in the upper extremities and severe lower extremity weakness, worse in the right leg, involving both proximal and distal muscles. All reflexes were absent, and mild distal sensory loss was present to all modalities in the lower extremities.
Investigations on admission to hospital included CSF examination which demonstrated a normal CSF protein and cell count. Other serological and hematologic tests were unremarkable. Nerve conduction studies showed a moderate to severe mixed axonal and demyelinating polyneuropathy. Because of the rapid progression and asymmetric features, the differential diagnostic possibilities were felt to include Guillain–Barre syndrome and vasculitic neuropathy. Sural nerve biopsy was performed.
Following a diagnosis of amiodarone neuromyopathy, the dosage was reduced to 200 mg/day, with subsequent complete resolution of the patient’s symptoms over a period of 3–6 months.
18.3 Disulfiram Neuropathy
18.3.1 Clinical Manifestations
Disulfiram (Antabuse) is used in the treatment of alcoholism, at a dose of 250–500 mg daily. Peripheral neuropathy occurs in a small fraction of patients and is probably dose dependent. Onset is usually within months after initiating therapy but can occur as soon as 10 days with high-dose treatment (van Rossum et al. 1984; Watson et al. 1980; Nukada and Pollock 1981). Distal symmetrical sensory and motor deficits are present. Some patients have also demonstrated evidence of central nervous system dysfunction (Kane 1970). Electrophysiological tests reveal an axonal neuropathy. Withdrawal of the drug usually results in substantial improvement over several months, although mild residual symptoms and electrophysiological abnormalities tend to persist.
18.3.2 Pathology
We have examined nerve biopsies from five patients with disulfiram neuropathy, and a substantial quantity of pathological material is available in the literature (Bilbao et al. 1984; Watson et al. 1980; van Rossum et al. 1984; Mokri et al. 1981; Moddel et al. 1978; Ansbacher et al. 1982; Nukada and Pollock 1981; Bergouignan et al. 1988; Bouldin et al. 1980). Active axonal degeneration is usually visible, with little or no segmental demyelination (Fig. 18.9a–d). Larger myelinated fibers may be preferentially involved, and unmyelinated fibers tend to be spared. In cases biopsied after cessation of the drug, regenerating clusters have been prominent (Moddel et al. 1978). One reported case has demonstrated a predominance of segmental demyelination which seemed to be independent of axonal disease (Nukada and Pollock 1981), and a rare onion-bulb formation was seen. We have never observed any changes suggesting a primary demyelinating component to this toxic neuropathy.
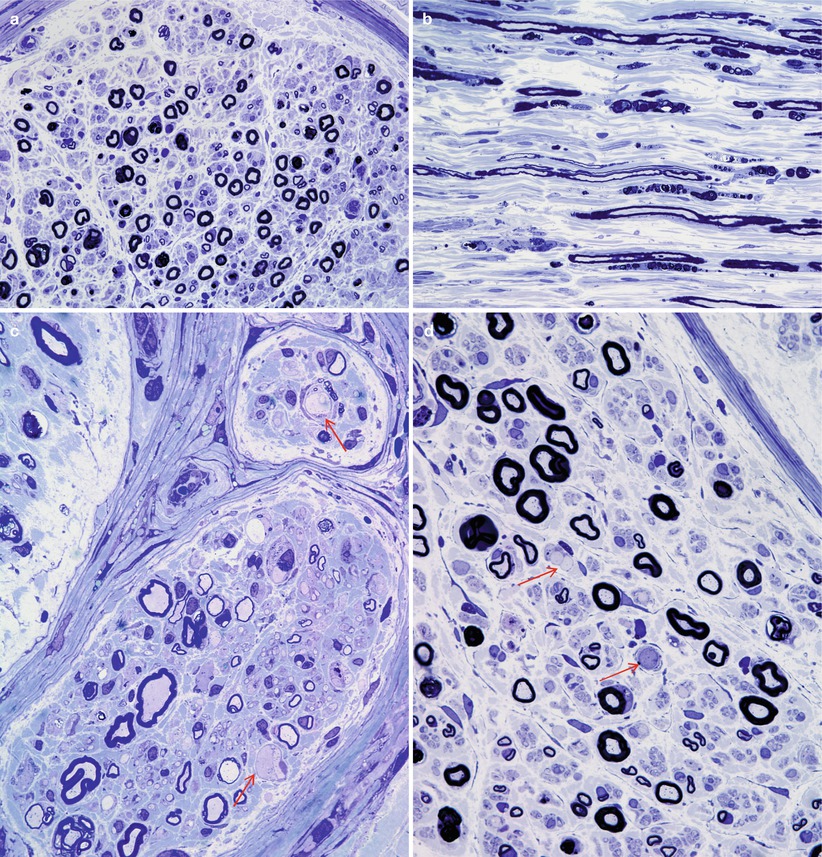
Fig. 18.9
Disulfiram: active axonal degeneration is seen against a backdrop of moderate axonal depletion (a, b). Several swollen axons are shown (arrows, c, d) (1 μm thick plastic sections; magnification: a, b 400×; c, d 1,000×)
Ultrastructural examination usually reveals a nonspecific pattern of active axonal degeneration with relative or absolute sparing of unmyelinated fibers. A minority of the reported cases (1 of 5 in our material) have demonstrated axoplasmic filamentous accumulations (see Chap. 5), producing a giant axonal change (Fig. 18.10a, b), which may be accompanied by a variety of accumulated organelles (Fig. 18.10c) (Bilbao et al. 1984; Bergouignan et al. 1988; Ansbacher et al. 1982).