Tumors of the Meninges
Thomas J. Kaley
INTRODUCTION
Diseases of the dura are rather uncommon, except for meningiomas. Not just the most common dural disease, meningiomas are the most common intracranial primary tumor in adults. Especially in the current environment of neuroimaging with computed tomography (CT) or magnetic resonance imaging (MRI) being performed for any and every possible neurologic symptom, both symptomatic and more frequently asymptomatic meningiomas will be found. Accordingly, meningiomas may become the most common intracranial tumor neurologists, and most physicians, will encounter. Although they are usually benign, they may enlarge and/or cause neurologic dysfunction depending on the location and because the cranium has only a limited capacity to accommodate an expanding mass. Surgery remains the treatment of choice, but not all meningiomas are amenable to complete resection, and some will recur and require further therapy. This chapter will outline the major dural diseases with the majority devoted to meningiomas.
MENINGIOMAS
EPIDEMIOLOGY
Meningiomas are the most common primary “brain” tumor in adults, although technically, they are tumors of the brain covering rather than brain parenchyma. The Central Brain Tumor Registry of the United States estimates an incidence of approximately 20,000 new meningioma cases diagnosed per year. This represents 36% of all primary brain tumors (World Health Organization [WHO]). These tumors have a clear female predominance with an almost 2:1 female-to-male ratio. These tumors are quite rare in children except within a predisposing neurogenetic syndrome. They are substantially more common in adults and tend to increase in incidence with age, particularly older than the age of 65 years. Autopsy series suggest a prevalence of 2%, typically asymptomatic and discovered incidentally during life or postmortem.
The only clear risk factors for meningiomas are prior radiotherapy involving the head and neck region as well as predisposing neurogenetic syndromes, such as neurofibromatosis type 2 (NF2). Ionizing radiation has also been demonstrated to be clear risk factor for future meningioma development. Prior radiation therapy for an extracranial malignancy such as head and neck cancer or childhood leukemia is the most common. These meningiomas that develop as a result of prior radiotherapy tend to be more aggressive in their clinical course. The latency for meningioma development is inversely proportional to the dose of radiation delivered.
Although no longer performed, scalp radiation for tinea capitis, a fungal infection, has been linked to meningioma development. Similarly, older less refined dental x-rays have been inconclusively linked to meningiomas. Currently, however, radiotherapy is no longer performed for scalp infections and newer dental x-rays probably do not have significant risk, different from the older less refined variant. Probably the most controversial potential link to meningiomas in the recent media and scientific literature has been the use of cellular phones and potential radiation exposure. To date, there has been no proven definitive link between cellular phones and the development of a meningioma.
PATHOBIOLOGY
Meningiomas are tumors that arise from the arachnoid coverings of the brain. Histologically, these tumors have various appearances depending on the subtype of meningioma, although most physicians frequently recall the whorling pattern of tumor cells which is most common and calcium collections known as psammoma bodies. The WHO classifies meningiomas into three grades (in the following text).
The most common neurogenetic syndrome predisposing the patient to meningiomas is NF2, the less common form of neurofibromatosis. NF2 is an autosomal dominant disorder with full penetrance caused by alteration of the NF2 gene (also known as chromosome 22, which encodes for the tumor suppressor protein merlin (moesin-ezrin-radixin-like protein), also called neurofibromin. Approximately 80% of sporadic meningiomas also harbor NF2 alterations. However, there is no clear selective therapy to target merlin alterations.
Recent research efforts have focused on understanding the biology of cancer as a whole and using targeted therapies that disrupt a specific biologic pathway, typically a growth factor ligand/receptor-mediated pathway. Meningiomas are highly vascular tumors with elevated expression of the vascular endothelial growth factor (VEGF) receptor and its ligand, VEGF. Other growth factor pathways such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), and somatostatin are also expressed in meningiomas. Efforts at targeting these pathways are discussed in the following text.
A major thrust of cancer investigation over the last decade has been whole genome sequencing to identify drugable oncogenic driver mutations. Mutations in two pathways recently were identified in approximately 20% of meningiomas: AKT and SMO. AKT mutations disrupt the homeostasis that normally regulates cancer cell survival, proliferation, angiogenesis, and metabolism. SMO mutations alter a key pathway responsible for controlling cancer cell self-renewal.
In the Clark et al. paper, genomic analysis revealed oncogenic mutations in AKT1 and SMO in 13% and 4% of specimens, respectively. These mutated tumors tend to occur more frequently in skull base locations, which also tend to be the most surgically challenging or inaccessible areas. Patients with AKT mutations harbored the E17K mutation, a well-characterized oncogenic mutation present in several other cancer types. The E17K mutation can be found in breast cancer, colorectal cancer, and lung cancer and results in constitutive AKT activation, which in turn stimulates downstream mTOR activity, resulting in overactivity of the PI3K pathway. PI3K pathway activation in turn stimulates multiple oncogenic processes including cell proliferation and angiogenesis. The SMO mutations included the L412F seen in a subset of medulloblastomas and the
W535L alteration seen in basal cell carcinoma and predictive of sensitivity to U.S. Food and Drug Administration (FDA)-approved therapy with vismodegib, a hedgehog inhibitor. When the SMO protein is activated, it results in ligand-independent downstream activation of the hedgehog pathway resulting in cell proliferation and cancer stem cell self-renewal.
W535L alteration seen in basal cell carcinoma and predictive of sensitivity to U.S. Food and Drug Administration (FDA)-approved therapy with vismodegib, a hedgehog inhibitor. When the SMO protein is activated, it results in ligand-independent downstream activation of the hedgehog pathway resulting in cell proliferation and cancer stem cell self-renewal.
In the Brastianos et al. paper, 65 meningioma specimens underwent analysis and revealed similar results identifying AKT and SMO mutations in a subset of patients. Five patients (8%) demonstrated AKT mutations, all E17K. Three patients (5%) demonstrated SMO mutations, two of which were the W535L mutation seen in basal cell carcinoma and one L412F alteration previously described in desmoplastic medulloblastoma. They then studied an additional 46 grade I tumors and 49 grade II and III tumors and found 6 AKT1 E17K mutated tumors (1 grade III, 5 grade I), and 2 SMO L412F mutated tumors (both grade I).
In summary, these two studies demonstrated that PI3K pathway activation and hedgehog pathway activation occurs in a subset of meningioma patients. The mutations seen are well characterized in other cancer types and in the case of basal cell cancer, predict therapeutic response to inhibition with an available agent. Future studies hope to define the role, if any, of these agents in the treatment of these particular molecular subtypes of meningioma.
CLINICAL MANIFESTATIONS
As with most other brain tumors, the clinical presentation of meningiomas is highly variable, depending on tumor location, size, and shape. Tumors may manifest as visual disturbances, speech dysfunction, weakness, sensory dysfunction, neurocognitive dysfunction, personality changes, or seizures. Because meningiomas are typically slow-growing neoplasms, they rarely cause acute increases in intracranial pressure, resulting in symptoms such as headache, nausea, vomiting, or alteration of consciousness. In fact, because they are such slow-growing tumors, they can often become surprisingly large without causing any clinical neurologic dysfunction. However, even a “benign” meningioma can result in disabling neurologic dysfunction if it grows beyond the capacity of the skull to accommodate the increase in volume. Rarely, these tumors may metastasize outside the central nervous system (CNS), usually to bone. This is an exceptionally rare event and typically only with malignant meningiomas.
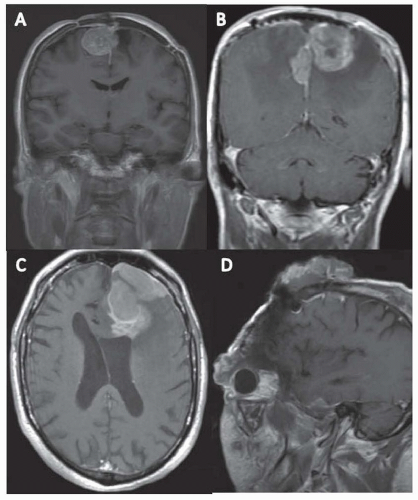 FIGURE 98.1 MRI appearance of meningiomas. A: Coronal T1 postcontrast image demonstrating a recurrent grade I meningioma. B: Coronal T1 postcontrast image demonstrating an atypical meningioma noting significant peritumoral edema. C: Axial T1 postcontrast image demonstrating an anaplastic meningioma noting brain invasion. D: Sagittal T1 postcontrast image demonstrating extracranial growth of a recurrent surgery and radiation refractory meningioma. |
DIAGNOSIS
Meningiomas are best visualized on an MRI with contrast, which demonstrates a brightly enhancing mass arising off of the dura which typically has a linear continuation with the dura known as a dural tail (Fig. 98.1). CT with contrast may demonstrate the same pattern of enhancement, but MRI provides a better visualization of the underlying brain. CT may be superior for demonstrating any changes in the neighboring bone, as meningiomas can trigger a reactive hyperostosis in the bone adjacent to the meningioma itself, seen in approximately 25% of patients. Without contrast, meningiomas appear on both CT and MRI as either hypodense or hypointense, or isodense or isointense.
As with most tumors, neuroimaging alone cannot definitively diagnose a meningioma, which depends on tissue analysis
acquired by biopsy or resection. The WHO classifies meningiomas in three grades (Table 98.1). WHO grade I meningiomas are classified as “benign” meningiomas and makeup approximately 80% of meningiomas. They are typically slow-growing tumors that basically fail to demonstrate any of the characteristics of higher grade meningiomas. Grade I meningiomas include most of the histologic subtypes: angiomatous, fibrous, lymphoplasmacyte-rich, meningothelial, metaplastic, microcystic, psammomatous, secretory, and transitional meningiomas. WHO grade II meningiomas include the clear cell and chordoid histologic variants, as well as atypical tumors that harbor increased mitotic activity as evidenced by four or more mitoses per 10 high-power fields or at least three of the following features: increased cellularity, small cells with a high nuclear-to-cytoplasmic ratio, prominent nucleoli, patternless or sheet-like growth, or necrosis. Importantly, brain invasion is associated with increased rates of recurrence and is sufficient to diagnose a WHO grade II tumor, regardless of the presence of other atypical features. WHO grade III meningiomas include rhabdoid, papillary, and anaplastic (malignant). The criteria for anaplastic meningiomas include increased mitotic activity as evidenced by 20 or more mitoses per 10 high-power fields and/or malignant characteristics resembling carcinoma, sarcoma, or melanoma, such as the loss of usual meningioma growth patterns, infiltration of underlying brain, abundant mitoses with atypical forms, and multifocal necrosis. Anaplasia is sufficient to diagnose a WHO grade III regardless of the presence of brain invasion.
acquired by biopsy or resection. The WHO classifies meningiomas in three grades (Table 98.1). WHO grade I meningiomas are classified as “benign” meningiomas and makeup approximately 80% of meningiomas. They are typically slow-growing tumors that basically fail to demonstrate any of the characteristics of higher grade meningiomas. Grade I meningiomas include most of the histologic subtypes: angiomatous, fibrous, lymphoplasmacyte-rich, meningothelial, metaplastic, microcystic, psammomatous, secretory, and transitional meningiomas. WHO grade II meningiomas include the clear cell and chordoid histologic variants, as well as atypical tumors that harbor increased mitotic activity as evidenced by four or more mitoses per 10 high-power fields or at least three of the following features: increased cellularity, small cells with a high nuclear-to-cytoplasmic ratio, prominent nucleoli, patternless or sheet-like growth, or necrosis. Importantly, brain invasion is associated with increased rates of recurrence and is sufficient to diagnose a WHO grade II tumor, regardless of the presence of other atypical features. WHO grade III meningiomas include rhabdoid, papillary, and anaplastic (malignant). The criteria for anaplastic meningiomas include increased mitotic activity as evidenced by 20 or more mitoses per 10 high-power fields and/or malignant characteristics resembling carcinoma, sarcoma, or melanoma, such as the loss of usual meningioma growth patterns, infiltration of underlying brain, abundant mitoses with atypical forms, and multifocal necrosis. Anaplasia is sufficient to diagnose a WHO grade III regardless of the presence of brain invasion.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


