and Robert E. Schmidt2
(1)
Sunnybrook and St Michael’s Hospitals, University of Toronto, Toronto, ON, Canada
(2)
Division of Neuropathology Department of Pathology, Washington University School of Medicine, St. Louis, MO, USA
Some polyneuropathies which have not been discussed elsewhere in this monograph deserve specific mention. Some of these are parts of clinical syndromes, while others are specific entities. Although mitochondrial diseases might well be considered to be “genetically determined,” they are also considered here. Peripheral neuropathy resulting from a mitochondrial defect associated with MFN2 and GDAP1 mutations has been addressed as part of the CMT group of peripheral nerve disorders and will not be readdressed here.
21.1 Neuropathy in Mitochondrial Diseases
21.1.1 Clinical Manifestations
Mitochondrial diseases are diagnosed when a disturbance of number, structure, or function of mitochondria is identified in association with an appropriate clinical picture. The spectrum of these diseases is growing rapidly. Mitochondria contain 2–10 copies of a 16,569 base-pair ring which encodes 13 proteins involved in oxidative phosphorylation and the special transfer RNA and ribosomal RNA required for intramitochondrial protein synthesis. Mitochondrial diseases usually arise as a consequence of mitochondrial DNA (mtDNA) mutations, but may also result from defects in nuclear DNA related to mitochondrial function. Although mitochondrial diseases may directly result in neuropathy, it is important to recognize that polyneuropathy may reflect a secondary associated condition (e.g., diabetes, renal failure, hypothyroidism) which may be part of some mitochondrial disorders (Finsterer 2005). Finsterer’s study (2005) of 108 patients with mitochondrial disease and polyneuropathy attributed 35 % directly to a primary mitochondrial pathogenesis.
Mitochondrial DNA defects are divided into deletions and point mutations (Zeviani and Antozzi 1992). Deletions are usually sporadic and typically manifest as a progressive external ophthalmoplegia (PEO) syndrome, either in isolation, with myopathy, or as the full Kearns–Sayre syndrome (KSS). Point mutations are usually inherited (typically maternal transmission) and are associated with a variety of clinical phenotypes including MELAS (myopathy, encephalopathy, lactic acidosis, stroke-like episodes), MERRF (myoclonus epilepsy with ragged red fibers), and Leber hereditary optic neuropathy. Peripheral neuropathy defines several mitochondrial syndromes: (1) NARP (neuropathy, ataxia, and retinitis pigmentosa) caused by a missense mutation in the mitochondrial ATPase gene (MTATP6, Holt et al. 1990), (2) MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) due to a thymidine phosphorylase (TYMP) gene defect, and (3) SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis) thought, in some patients, to be caused by mutations in the mitochondrial polymerase γ (POLG) gene (Van Goethem et al. 2003). MNGIE is a particularly interesting neuropathy in which a nuclear-encoded defect in endothelial cell growth factor 1 (ECGF1) results in secondary mutations, deletions, and defects in mtDNA. Neuropathy has also been described in a variety of other mitochondrial syndromes, including MELAS, MERFF, PEO, and KSS (Nardin and Johns 2001). Leigh subacute necrotizing encephalopathy has been associated with several different mtDNA and nuclear DNA defects, illustrating the potential genetic heterogeneity of mitochondrial disease syndromes.
Neuropathy is present in 25–50 % of patients with most mitochondrial diseases (Eymard et al. 1991; Mizusawa et al. 1991; Yiannikas et al. 1986) and is usually mild, length dependent, with sensory alterations often predominating over motor deficits. Nerve conduction studies show axonal neuropathy, but demyelinating features have also been reported (Finsterer 2005). Peripheral neuropathy may occur (1) in mitochondrial disorders associated with defects in mitochondrial DNA maintenance or replication which may be caused by nuclear gene mutations (POLG1, C10ORF2, TYMP, and MPV17), (2) in the respiratory chain complexes particularly complex V associated with MTATP6 mutations, and (3) in mitochondrial dynamics, i.e., the distribution of mitochondria by fission and fusion (MFN2, GDAP1 mutations) (Pareyson et al. 2013). A sense of the current complexity of mitochondriopathy in association with peripheral neuropathy is provided by the recent review by Pareyson and colleagues (2013).
Recent studies using mutant mice lacking Schwann cell Tfam, which controls the transcription, replication, and stability of mtDNA, show neuropathy characterized by the depletion of mtDNA, mitochondrial morphologic abnormalities, failure to create a normal electron transport chain, and respiration deficiency (Viader et al. 2013). In this mouse model, neuropathy was characterized by a preferential loss of small unmyelinated C fibers, followed by that of large myelinated axons and, ultimately, extensive demyelination of residual axons (Viader et al. 2013).
21.1.2 Pathology
Many single cases and small series of nerve biopsies and autopsies have been recorded in mitochondrial diseases including PEO/myopathy (Drachman 1968; Gemignami et al. 1982; Gonatas 1967; Markesbery 1979; Mizusawa et al. 1991; Peyronnard et al. 1980; Pezeshkpour et al. 1987; Schroder and Sommer 1991; Yiannikas et al. 1986), Leigh disease (Goebel et al. 1986; Jacobs et al. 1990; Schroder and Sommer 1991), MERRF (Mizusawa et al. 1991; Sasaki et al. 1983), MELAS (Mizusawa et al. 1991; Nicoll et al. 1993; Schroder and Sommer 1991), and MNGIE (Bardosi et al. 1987; Hirano et al. 1994, 2004; Li et al. 1991; Simon et al. 1990; Steiner et al. 1987; Threkeld et al. 1992; Uncini et al. 1993). As discussed below, the histological picture is nonspecific and does not permit differentiation between the various mitochondrial diseases.
21.1.2.1 Light Microscopy
Nerve biopsies and autopsy reports in patients with PEO/myopathy syndrome have almost invariably revealed chronic depletion of myelinated fibers, the larger MFs usually being more severely affected. In one report, the study of myelin lamellar numbers vs. axonal diameter suggested the presence of axonal atrophy (Pezeshkpour et al. 1987). Regenerating clusters may be increased (Mizusawa et al. 1991; Peyronnard et al. 1980). Unmyelinated fibers are relatively spared, although observations such as increased numbers of denervated bands or collagen pockets suggest a mild involvement (Pezeshkpour et al. 1987; Yiannikas et al. 1986). Myelin alterations such as hypomyelination (Peyronnard et al. 1980), and segmental demyelination and remyelination with small onion-bulb (OB) formations (Mizusawa et al. 1991), have been frequently noted, but whether these are primary or secondary to axonal alterations has not been explored. The dramatic hypertrophic neuropathy described in Drachman’s “Ophthalmoplegia Plus” paper (Drachman 1968) has not been described by other workers, is clinically and histologically atypical for a mitochondrial disease, and most likely represents a superimposed peripheral nerve condition, perhaps multifocal neuropathy with persistent conduction block or CIDP.
Study of peripheral nerve in patients with Leigh disease has indicated preservation of myelinated and unmyelinated fiber densities, with thinly myelinated axons resulting from demyelination and remyelination (Goebel et al. 1986), or intrinsic hypomyelination (Jacobs et al. 1990; Schroder 1993). “Incipient” onion bulbs in Leigh disease were described by Goebel et al. (1986). Wallerian degeneration, active segmental demyelination, and dominant onion-bulb formation have not been noted.
In the point mutation syndromes, MELAS and MERRF, biopsies and autopsy studies have revealed chronic axonal depletion with some thinly myelinated fibers and occasional onion bulbs (Mizusawa et al. 1991; Nicoll et al. 1993; Robitaille et al. 1980). Sural nerve biopsies in patients with SANDO demonstrate severe loss of both large and small myelinated fibers with endoneurial fibrosis (Fadic et al. 1997).
Light microscopy in the MNGIE syndrome has also revealed chronic axonal depletion with variable regenerative activity and evidence of demyelination and remyelination. Dystrophic mitochondria with loss of cristae and paracrystalline inclusions have been described (Pareyson et al. 2013). The symptoms of MNGIE are an example of a mitochondrial syndrome caused by an mtDNA replication dysfunction that results from an imbalance in the mitochondrial nucleotide pool. Most often some small onion bulbs are seen, with individual reports describing a spectrum ranging from none to numerous onion bulbs. Whether the myelin alterations are primary or secondary remains unclear. Various workers have commented on nonspecific findings such as endoneurial vascular hyalinization and perineurial or endoneurial fibrosis.
21.1.2.2 Electron Microscopy
Of interest as to pathogenesis of the neuropathy is whether abnormal mitochondrial morphology can be detected in peripheral nerve as readily as it is in the muscle. Increased numbers or focal accumulations of mitochondria have occasionally been reported in Schwann cells (Peyronnard et al. 1980; Threkeld et al. 1992), and we have encountered an example in a case of neuropathy with genetically unestablished mitochondrial encephalomyopathy (Fig. 21.1a–c). Similar features were noted in neural endothelial and vascular smooth muscle cells in KSS and MELAS (Schroder and Sommer 1991), as previously described in other tissues (Ohama et al. 1987). Enlarged mitochondria have also been described in these mitochondrial neuropathies (Goebel et al. 1986; Hirano et al. 1994). Schroder and Sommer (1991) have studied this issue most thoroughly and have concluded that enlarged mitochondria, some containing distorted cristae, are more common in mitochondrial cytopathies than in other neuropathies, but are not specific. Recent studies of a variety of a large number of mitochondrial peripheral neuropathies (Vital and Vital 2012) confirm the presence of ultrastructural mitochondrial changes including enlarged mitochondria with intertwined, or amputated cristae in Schwann cell cytoplasm. Increased numbers of Reich Pi granules have been emphasized in some reports (Gemignami et al. 1982; Gonatas 1967).
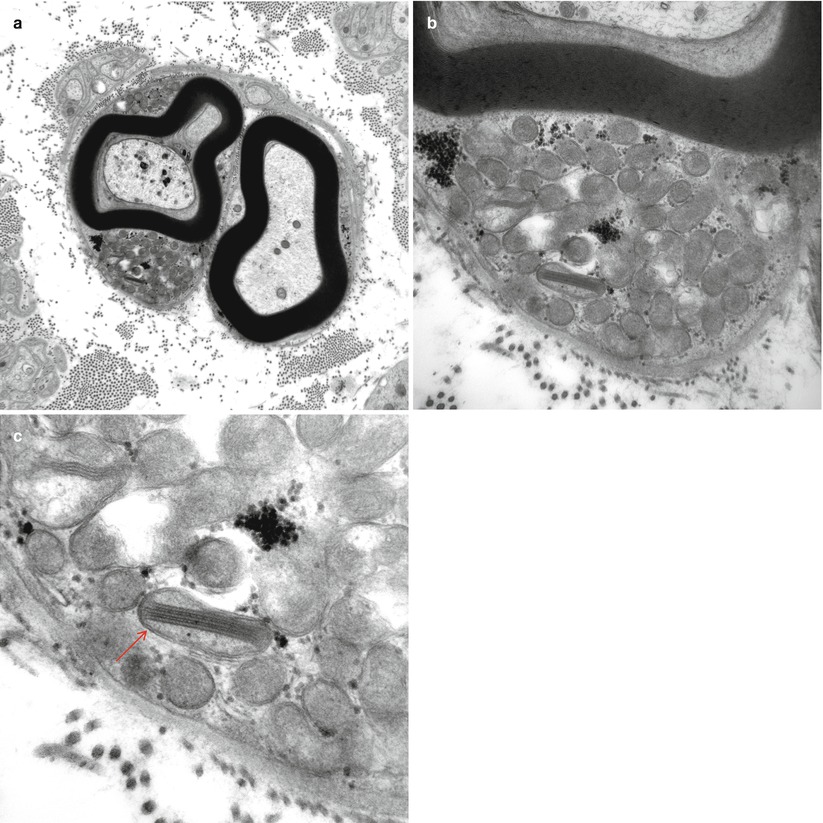
Fig. 21.1
Sural nerve in a patient with unestablished mitochondrial disease. Note that the Schwann cell collections of mitochondria (a, b) demonstrate one with a crystalline inclusion (arrow, c) (magnification: (a) 12,000×; (b), 32,000×; (c), 72,000×)
A number of authors have described and illustrated nonmyelinating Schwann cells containing paracrystalline material surrounded by a double membrane, likely of mitochondrial origin (Schroder 1993; Yiannikas et al. 1986). However, such inclusions are not infrequently seen in a variety of non-mitochondrial peripheral nerve diseases (Schroder 1993).
Classic inclusions of the paracrystalline stacked bar variety have been illustrated in the nerve in only one instance (Li et al. 1991). Other, less characteristic but unusual intramitochondrial structures have been infrequently noted (Goebel et al. 1986; Pezeshkpour et al. 1987). In two patients with CMT type 6 and another with an idiopathic sensorimotor neuropathy, Sommer and Schroder (1989) illustrated enlarged mitochondria in Schwann cells of small MFs containing several linearly arrayed and closely apposed cristae with a paracrystalline material filling the intercristal space. However, the “mitochondrial” nature of some CMT-6 patients remains speculative pending further evidence of mitochondrial dysfunction in this rare syndrome.
In one report of a patient with PEO/myopathy, examination of intramuscular nerve fibers revealed intra-axonal structures identical in ultrastructural appearance to Hirano bodies (Atsumi et al. 1980).
21.2 Adult Polyglucosan Body Disease
21.2.1 Clinical Manifestations
A number of entities are associated with tissue polyglucosan bodies, including type IV glycogenosis, phosphofructokinase deficiency, Lafora body disease, and normal aging (Cavanagh 1999). In our experience, polyglucosan bodies are found in about 3 % of sural nerve biopsies. In recent years, adult polyglucosan body disease (APGBD) has emerged as another distinct entity associated with characteristic inclusions (Cafferty et al. 1991; Gray et al. 1988; Karpati and Carpenter 1983; McDonald et al. 1993; Robitaille et al. 1980; Mochel et al. 2012). The APBGD syndrome includes adult-onset progressive upper motor neuron disease, urinary incontinence, dementia, and sensorimotor or pure motor peripheral neuropathy. Family history is positive in about one-third of cases, in a pattern suggestive of autosomal recessive inheritance. APGBD (Phenotype MIM#263570) is caused by mutation in the glycogen branching enzyme gene (GBE1; Gene/Locus MIM#607839). However, mutation in the same gene (GBE1) causes type IV glycogen storage disease (GSD IV; Phenotype MIM#232500), an early childhood disorder with systemic manifestations. Polyglucosan bodies are found throughout the central and peripheral nervous systems, as well as in myocardium and other nonneural tissues (Gray et al. 1988; Robitaille et al. 1980). Electrophysiological testing demonstrates an axonal neuropathy.
21.2.2 Pathology
A nerve biopsy showing polyglucosan bodies (PGBs) is commonly employed for the diagnosis of APGBD in life, although axillary skin biopsy may be a reliable and less invasive procedure, showing large numbers of PGBs in the myoepithelial cells of apocrine glands (Busard et al. 1991; Milde et al. 2001).
Nerve biopsies reported in APGBD have shown from one to many polyglucosan bodies (Fig. 21.2a–c) per nerve fascicle cross section. Typically the bodies are intra-axonal, round, and range from 5 to 70 μm in diameter (Fig. 21.2a–c), may result in minimal change in the axon/myelin (Fig. 21.2d), or induce expansion of the axon dimensions with myelin thinning (Fig. 21.2e). Polyglucosan bodies are composed of aggregates of 6–8 nm diameter branched filaments (Fig. 21.2f) which lack a limiting membrane. The inclusions are seen in myelinated axons and, less frequently, in unmyelinated axons or Schwann cells (Cafferty et al. 1991; Vos et al. 1983). Longitudinal sections may indicate that the inclusions are elongated, and several may be seen within one axon (Cafferty et al. 1991; Robitaille et al. 1980). Deposits of similarly staining, but less distinctively organized, material can be seen in axons or other peripheral nerve cells including endothelial and perineurial cells, fibroblasts, and macrophages (Vos et al. 1983). Extracellular PGBs, surrounded by inflammatory cells, may result from the recent death of the axon previously containing that inclusion (Robitaille et al. 1980).
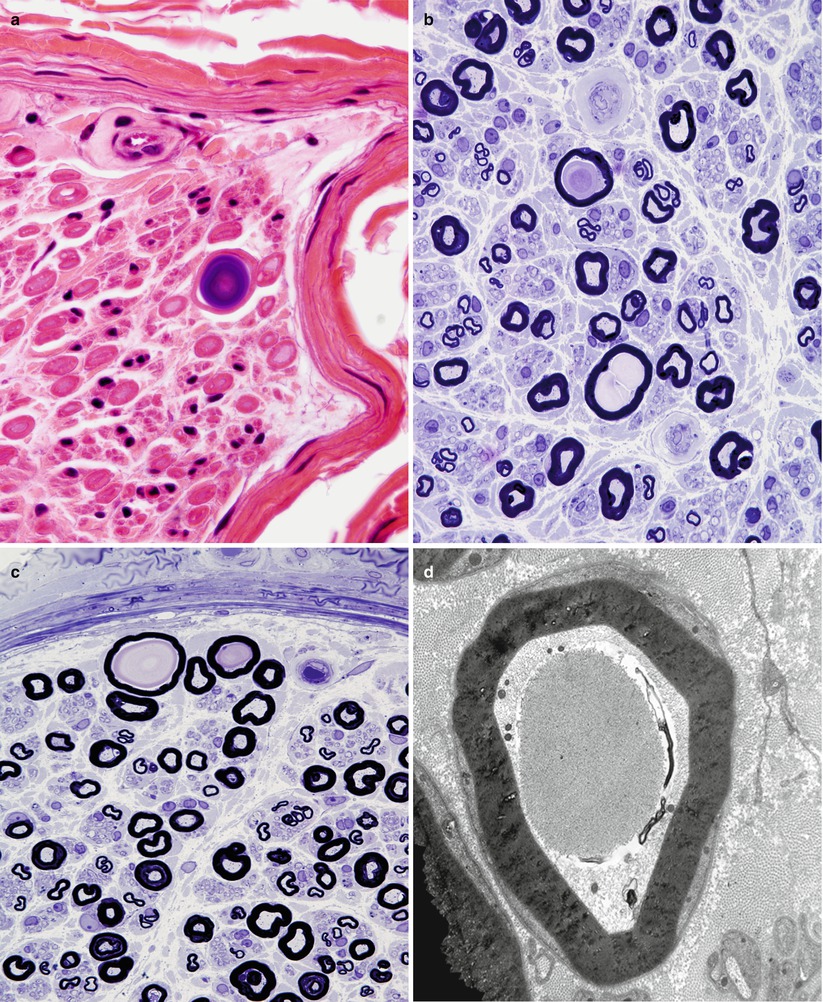
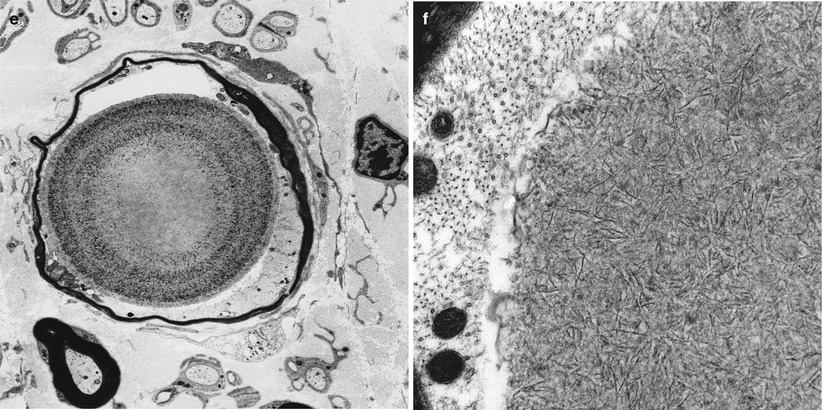
Fig. 21.2
Sural nerve in a 65-year-old patient with vasculitis. In the older age group, rare polyglucosan bodies (a–c) are of undetermined significance in the absence of an appropriate clinical context. Ultrastructural analysis demonstrates intra-axonal polyglucosan bodies (d) which may dilate the axon and thin the myelin sheath (e). Ultrastructural analysis shows filamentous aggregates which are not membrane delimited (f) (a, H&E; b, c 1 μm thick plastic sections; d–f)
In most reported cases of APGBD axonal dropout ranging from mild to severe has been the dominant finding, the infrequency of debris-filled macrophages indicating a slow degenerative process. Regenerating clusters may be present. Prominent segmental demyelination is less commonly seen (Matsumuro et al. 1993; Vos et al. 1983). Axons with large inclusion bodies can show attenuation of surrounding myelin, but this may be due to axonal dilation, not demyelination and remyelination (Yoshikawa et al. 1990).
21.2.3 Pathogenesis
Polyglucosan bodies (including Lafora bodies and corpora amylacea) are made up almost entirely by glucose polymers, with only a small amount of protein, and some sulfate and phosphate groups (Stam and Roukema 1973; Steyaert et al. 1990; Milde et al. 2001). A study in alloxan diabetic rats demonstrated an intimate relationship between glycogen accumulation and formation of polyglucosan bodies (Powell et al. 1979). Seemingly identical inclusions are seen in type IV (brancher deficiency) glycogenosis (McMaster et al. 1979; Schroder et al. 1993). Thus, it seems reasonable to suppose that a disturbance of carbohydrate metabolism might result in the production of PGBs. Indeed, several Ashkenazi Jewish patients with APGBD have been shown to have a tissue-specific defect of brancher enzyme (Bruno et al. 1992; Lossos et al. 1991).
The mechanism of production of the neuropathy is unclear. Polyglucosan bodies are most commonly seen in the presence of an axonopathy (Busard et al. 1990); however, the axon is not necessarily disrupted by the inclusions, and in fact seems to be able to adapt to their presence (Yoshikawa et al. 1990). Whether the polyglucosan bodies, when massive in size or present in large numbers, cause the neuropathy or are an epiphenomenon of the metabolic defect that underlies the neuropathy remains unknown.
21.2.4 Differential Diagnosis
Our experience suggests that one or two polyglucosan bodies seen in a nerve biopsy specimen are a nonspecific finding with no diagnostic implications. We have noted polyglucosan bodies in 2–3 % of sural nerve biopsies, and others have reported that 8–15 % of unselected sural nerve biopsies in adults may show this finding (Averback and Langevin 1978; Busard et al. 1990). Intramuscular nerve twigs show polyglucosan bodies even more frequently (Averback and Langevin 1978; Bernsen et al. 1989; Fukuhara 1977). The incidence of PGBs in the nerve clearly rises with age, as with corpora amylacea in the central nervous system (Averback and Langevin 1978; Bernsen et al. 1989; Busard et al. 1990; Thomas et al. 1980), and there may be an association between the presence of nonspecific CNS disease and the finding of peripheral nerve PGBs, even if a syndrome suggestive of APGBD is absent (Busard et al. 1990).
Observation of more than one PGB per nerve fascicle, of a PGB outside an axon, or of unusually large PGBs (>30 μm) should, however, lead to consideration of a number of disease entities, including APGBD, type IV glycogenosis (McMaster et al. 1979; Schroder and Sommer 1991), and Lafora disease (Bernsen et al. 1989; Busard et al. 1990). Similar suspicions should be raised by the finding of even a single PGB in a patient under the age of 5 (Busard et al. 1990) and perhaps under the age of 20 (Bernsen et al. 1989). PGBs have also been described in the setting of CIDP (Busard et al. 1990; Yoshikawa et al. 1990) and neuropathy in a diabetic (Mancardi et al. 1985). One patient with numerous PGBs in peripheral nerve and clinical features suggestive of APGBD who came to autopsy failed to reveal any unusual pathological features suggestive of a systemic accumulation of polyglucosan bodies (Gray et al. 1988).
21.3 Sensory Neuropathy Syndrome
21.3.1 Clinical Manifestations
The differential diagnosis of sensory neuropathy is considerable (Table 21.1). Once systemic malignancy and identifiable neuropathies are excluded, there remain a considerable number of patients in whom no explanation can be given and who have a relatively characteristic syndrome (Dalakas 1986; Griffin et al. 1990; Sobue et al. 1993; Windebank et al. 1990). These patients develop paresthesias, sensory loss, and sensory ataxia, but show no significant weakness. Sensory abnormalities may be distal and symmetrical, asymmetric, or segmental and can include the cranial nerves. As suggested by the sensory ataxia, large fiber damage often predominates, and small fiber function may be strikingly spared. Rate of progression varies widely, from days to years, and the morbidity ranges from insignificant to debilitation. Despite an extensive search, investigations reveal no evidence of malignancy, as confirmed with long-term follow-up (Windebank et al. 1990). There is an association with collagen disease, especially Sjögren syndrome (Griffin et al. 1990), but this accounts for a minority of patients (Dalakas 1986; Sobue et al. 1993; Windebank et al. 1990). CSF protein is normal or minimally elevated, and cell count is normal. Nerve conduction abnormalities suggest sensory axon damage with and minimal or no involvement of motor axons. No therapy has been of benefit, but a small proportion of patients show some spontaneous improvement (Dalakas 1986; Windebank et al. 1990).
Table 21.1
Miscellaneous neuropathies
Neuropathy associated with mitochondrial disease |
Adult polyglucosan body disease |
Idiopathic sensory neuropathy/ganglionopathy |
Hypereosinophilic syndrome |
“Motor neuron” diseases |
Critical illness polyneuropathy |
Multiple symmetrical lipomatosis (Madelung disease) |
21.3.2 Pathology
Sural nerve biopsy has been performed in many of these patients (Dalakas 1986; Griffin et al. 1990; Sobue et al. 1993; Windebank et al. 1990). Inflammation is not a feature, except in patients with Sjögren syndrome who may demonstrate perivascular mononuclear cell collections. Axon loss ranges from minimal to severe, and status ranging from very active to indolent. Regenerating clusters are infrequent. Large myelinated fibers may be most involved, but small myelinated and unmyelinated axons can also be lost (Sobue et al. 1993). Evidence of segmental demyelination and remyelination on teased fiber preparations has been interpreted as secondary to axonal atrophy (Sobue et al. 1993; Windebank et al. 1990).
The sural nerve findings in idiopathic sensory ganglionopathy are entirely nonspecific and cannot be distinguished from those in the paraneoplastic variant, although prominent inflammatory infiltration should suggest the presence of the Sjögren syndrome. Other potential causes of a sensory neuropathy (Table 21.1), especially vasculitis and CIDP, should be looked for.
21.3.3 Pathogenesis
A small number of autopsies and dorsal root ganglion biopsies have demonstrated inflammation and cell body loss in the spinal ganglia (Griffin et al. 1990). However, spinal ganglionitis would not explain the occasional evidence of mild motor fiber involvement (Windebank et al. 1990). Despite the association with autoimmune diseases, especially Sjögren syndrome, circulating antibodies reactive against spinal ganglion neurons have not been found (Griffin et al. 1990), in contrast to paraneoplastic polyganglionopathy. Inflammation may be seen in proximal nerve trunks, as contrasted with the absence of inflammation in nerve biopsy specimens (Sobue et al. 1993). The sural nerve histological findings described above suggest that a “sensory variant” of CIDP is not an important cause of this syndrome (Windebank et al. 1990).
21.4 Neuropathy in the Hypereosinophilic Syndrome
21.4.1 Clinical Manifestations
The hypereosinophilic syndrome (HES) is characterized by peripheral blood eosinophilia in the absence of known associated disease such as parasitic or fungal infection, granulomatous angiitis, recognized allergic reaction, or malignancy (Moore et al. 1985). The clinical picture is of a multisystem disease with fevers, skin rash, pulmonary infiltrates, and cardiac disease (Chusid et al. 1975). Neuropathy may occur in as many as 50 % of patients, most often a mild symmetrical sensory polyneuropathy (Mondelli et al. 1993). However, a severe rapidly evolving axonal neuropathy, with distal predominance and some asymmetry, has been described as well (Dorfman et al. 1983; Grisold and Jellinger 1985; Sunohara et al. 1989). Response to steroids is usually gratifying.
21.4.2 Pathology
Nerve biopsy has been reported by several authors (Dorfman et al. 1983; Grisold and Jellinger 1985; Lupo et al. 1989; Monaco et al. 1988; Sunohara et al. 1989; Werner and Wolf 1990; Wichman et al. 1985). Most often a severe acute or chronic axonal neuropathy with little or no regenerative activity has been demonstrated. Large myelinated fibers may be more severely depleted. Biopsies in mildly affected or asymptomatic patients have shown only mild axonal loss or even a normal nerve (Vos et al. 1983; Wichman et al. 1985). Despite the usual presence of a marked peripheral eosinophilia, only one report describes an inflammatory neuropathy with significant vascular injury, and this case may have been an example of allergic eosinophilia, not an idiopathic HES (Grisold and Jellinger 1985). Endoneurial edema was prominent in the two cases described by Monaco et al. (1988). In some reports, the authors have been impressed by the prominence of degranulated mast cells or thickening of capillary walls and endothelial cells (Lupo et al. 1989; Monaco et al. 1988).
21.4.3 Pathogenesis
Many of the manifestations of the HES are reminiscent of Churg–Strauss angiitis, and the possibility that this is a clinical variant must be considered. The neuropathy in HES is often asymmetric, suggestive of ischemic origin. However, most nerve biopsy material in the HES has shown a noninflammatory axonal neuropathy, which differs from the necrotizing angiitis of Churg–Strauss. Ischemia may be due to eosinophil-induced endothelial cell injury (Dorfman et al. 1983). A possible role of axonotoxic eosinophil products has been postulated and receives some support from experimental data (Lupo et al. 1989; Sunohara et al. 1989).
21.5 Motor Neuron Diseases
The motor neuron diseases, including ALS and spinal muscular atrophy, are generally said to spare sensory fibers, and as such one would expect sensory nerve biopsy to be normal. However, the literature suggests that this is an erroneous assumption and that although motor abnormalities completely overshadow the sensory changes, the latter do occur (Dyck et al. 1975; Mondelli et al. 1993).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


