Fig. 2.1
Roles of astrocytes in the brain
Both astrocytes and microglia play a major role in regulation of neuroinflammation. Microglia invade the brain early in development and take on a resting ‘protective’ role as sentinels, scattered uniformly throughout the CNS and forming a network of potential effector cells. Astrocytes , which outnumber microglia within the CNS parenchyma, are the major components of the CNS innate immune system. Astrocytes have been reported to suppress T helper 1 (Th1) and T helper 2 (Th2) cell activation, the proliferation and effector functions of activated T cells, and possess a wide variety of molecular mechanisms to induce apoptosis in activated T cells (Amor et al. 2010). Activated astrocytes express an array of inflammatory cytokines and chemokines (Dong and Benveniste 2001). In addition to production of pro-inflammatory mediators, the stimulation of cultured astrocytes or cell lines results in expression of major histocompatibility complex (MHC) class II molecules and co-stimulatory molecules such as B7-1 and B7-2 (Soos et al. 1999). Although, earlier studies indicate that neurons play a passive role in inflammation, but recent studies indicate that neurons contribute to inflammation by providing many of their products (i.e. neuropeptides and transmitters), as well as the neuronal membrane proteins CD22, CD47, CD200, CX3CL1 (fractalkine), intercellular adhesion molecule (ICAM)-5, neural cell adhesion molecule (NCAM), semaphorins and C-type lectins. All these neuronal factors regulate neuroinflammation (Tian et al. 2009). In addition, neurons express low levels of major histocompatibility complex (MHC) molecules and actively promote T-cell apoptosis via the Fas–Fas ligand pathway (CD95–CD95 L). Accumulating evidence suggests that neurons use a variety of signals to modulate microglial cells and astrocytes . These signals can be categorized into excitatory and inhibitory signals. Inhibitory signals from neurons constitutively maintain microglial cells and astrocytes in their quiescent state and antagonize proinflammatory activity, whereas excitatory signals are inducible and incite activation of microglial cells and astrocytes under pathological conditions towards a beneficial or detrimental phenotype. Thus, various neuronal signaling molecules actively modulate microglial functions and contribute to the inflammatory milieu in neurodegenerative diseases (Biber et al. 2007; Farooqui 2010). Neurons also favor the differentiation of T-regulatory cells, by providing a local microenvironment dominated by transforming growth factor–β1 (TGF-β1) (Amor et al. 2010). Collective evidence suggest that communication among neurons, microglia, and astrocytes is essential in maintaining homeostasis in the brain as well as responding appropriately to a variety of neuroimmune challenges. Physiological, morphological, and functional alterations in neurons and microglia during aging, stress, and inflammation disrupt the normal cross-talk among these cells in resulting in a dysregulated neuroimmune environment with potential deleterious consequences on brain function and behavior. Although, increase in microglia activation and neuronal injury can be the result of an exaggerated neuroimmune responses, but it remains unknown if microglial overactivation precedes and causes neuronal damage, or if activation occurs in response to loss of normal neuronal integrity. Injured neurons trigger glial activation, resulting in the production of inflammatory molecules and phagocytosis of injured neurons by glial cells. On the other hand, these neurons can also suppress glial activation through the induction of anti-inflammatory cytokines and chemokines. It is proposed that astrocytes and microglia interact with neurons at the synapse to modulate synaptic function and plasticity (Eroglu and Barres 2010), and are also vital for host defense mechanisms and response to stress (Ransohoff and Perry 2009).
2.2 Contribution of Microglial Cells in Neuroinflammation
Microglial cells are resident macrophages of the brain . They account for 10–12 % of the total glial cell population in the brain. They are predominately found in the grey matter, with especially high concentrations in the hippocampus, hypothalamus, basal ganglia and substantia nigra (Block et al. 2007; Mittelbronn et al. 2001). They originate from yolk sac and invade the brain tissue during early embryonic development and proliferate locally in the brain (Ginhoux et al. 2010; Schulz et al. 2012). In contrast to other yolk sac-derived macrophages, they are not replaced during the postnatal period and later life by liver- or bone marrow-derived macrophages (Hoeffel et al. 2012). The total number and density of microglia have been shown to increase significantly with age in various regions of the brain, including the hippocampus (Mouton et al. 2002), visual and auditory cortices (Tremblay et al. 2010, 2012), and the retina (Damani et al. 2011). Under normal physiological conditions microglial cells have a small, somewhat elongated cell body with long, fine processes. The ramified microglial cells are rather evenly spaced throughout the brain, with their processes pervading the entire brain. It is generally accepted that ramified microglia constantly survey the CNS and synapses for intruders/stressors which may disrupt structure and function of neuronal circuits (Wake et al. 2009) . Microglia function is crucial for the homeostasis of the brain in health and disease, as they represent the first line of defense against pathogens and injuries, contributing to immune responses, but are also involved in tissue repair and remodeling (Lindsey et al. 1979). Toll-like receptors (TLRs) are first-line molecules for initiating innate immune responses (Akira and Takeda 2004). When activated through TLR signaling, microglial cells respond to injury and damaged neuronal cells by secreting chemokines and cytokines and express co-stimulatory molecules needed for protective immune responses to pathogens and efficient clearance of damaged tissues (Takeuchi and Akira 2010). Recent studies have indicated that distinct functional microglial phenotypes, ranging from the so-called M1-like proinflammatory to the M2-like antiinflammatory, can affect differently the health of mature, pre-existing neurons and the fate of neural stem progenitor cells (Ekdahl et al. 2009). The balance between pro- and antiinflammatory functions of microglia has been reported to affect the outcome of neuroinflammatory and regenerative and reparative mechanisms (Minghetti et al. 2005; Ekdahl et al. 2009) . Microglial cell activation is characterized by conspicuous changes in their ramified morphology to an intermediate and amoeboid forms resulting in round morphological profile of full phagocytes (Morioka et al. 1993; Thored et al. 2009). The morphological changes are accompanied not only by the upregulation of nuclear factor-κB (NF-κB) in cytoplasm and increase in expression of MHC classes I and II, complement C3, Fc, thrombin, scavenger receptors (i.e., CD36, SR-A, CD204, SR-BI), cytokine, chemokine in the nucleus. In addition, CD4 and CD8 receptors, toll-like receptors, P2X7 purinergic receptor, and several oxidative enzymes, such as NADPH oxidase (Streit et al. 1999; Husemann et al. 2002; Block et al. 2007; Ransohoff and Perry 2009; Helmut et al. 2011) are also increased. Activation of P2X7 purinergic receptor and NADPH oxidase induces the production of the superoxide, in primary rat microglia (Fig. 2.2) .
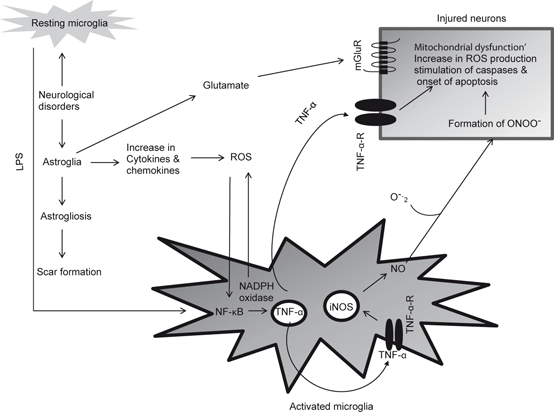
Fig. 2.2
Neurochemical changes associated with the activation of microglial cells and interactions among activated microglia, astroglia, and neurons. Lipopolyscharide (LPS), Reactive oxygen species (ROS), nuclear factor kappa-B (NF-κB), tumor necrosis factor-α (TNF-α), tumor necrosis factor-α receptor (TNF-α-R), inducible nitric oxide synthase (iNOS), nitric oxide (NO), superoxide (O 2 −), and peroxynitrite (ONOO −). The free radical NO reacts with O2 − to produce peroxynitrite (ONOO −), a powerful oxidant, which decomposes to form hydroxyl radical (HO ●)
The occurrence of activated microglial cells and enhancement of superoxides production have been reported in various brain regions (hippocampus, substantia nigra and spinal cord) of stroke, AD, PD, and ALS patients. The involvement of microglial cells in the pathogenesis of stroke, AD, PD, and ALS is supported by studies in animal and cell culture models of neurodegenerative diseases. It is shown that (a) microglial cell activation precedes the neurodegenerative changes; (b) activated microglial cells surround the region that undergo neurodegeneration and phagocytose the degenerating cells; (c) activated microglia not only release neurotoxic molecules such as interleukin (IL)-1β, IL-6, TNF-α , glutamate, aspartate, and quinolinic acid, nitric oxide, and reactive oxygen species (ROS) (Fig. 2.2), but also facilitate the assembly and activation of so called “inflammasomes” a cytoplasmic caspase-1 activating and self-oligomerizing signaling complex with molecular mass of greater than 700 kDa (Chakraborty et al. 2010); (d) inhibition of microglial activation results in the amelioration of neurodegeneration, and (e) microglia derived from aged animal exert more toxicity to neurons in an age-dependent fashion, in the same way neurodegenerative disorders occur. The release of interleukin (IL)-1β, IL-6, TNF-α leads to marked enhancement in activities of PLA2, COX-2, and 5-LOX and generation of proinflammatory eicosanoids and platelet activating factor (Phillis et al. 2006) (Fig. 2.3) . These pro-inflammatory mediators along with proteinases, and complement proteins intensify neuroinflammation. At this time it is difficult to establish the correct sequence of these events, so it is not clear whether activation of microglia and the associated inflammatory changes play a part in triggering neurodegenerative processes or whether cell activation is a response to the early changes associated with neurotraumatic and neurodegenerative diseases. In brain activated microglial cells not only express receptors for neurotransmitters such as ATP, adenosine, glutamate, GABA, acetylcholine, dopamine and adrenaline (Lee 2013), but also secrete a variety of immune system modulators including complement proteins, adhesion molecules, colony-stimulating factor-1, tumor and growth factors (TGF- α and β), monocyte chemotactic protein (MCP-1), and macrophage inflammatory peptide-1α (MIP-1α) (Minghetti et al. 2005; Galimberti et al. 2006; Janelsins et al. 2005). MCP-1 is a major chemokine that in cardiovascular and cerebrovascular systems attracts more monocytes to the plaque to enhance the inflammation. MCP-1 is abundantly expressed in atherosclerotic arterial lesions. The chemotactic response of the mononuclear cells is dependent on the presence of the chemokine receptor-2 (CCR-2) on its surface (Charo and Peters 2003) .
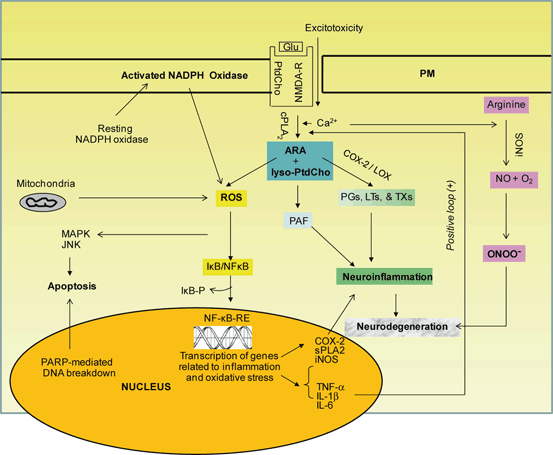
Fig. 2.3
Generation of reactive oxygen species (ROS), activation of NF-κB, and expression of cytokines. Cytokines stimulate PLA2, COX-2, and iNOS and contribute to the generation of PGs, LTs, TXs, and PAF. Phosphatidylcholine (PtdCho), lyso-phosphatidylcholine (lyso-PtdCho), arachidonic acid (ARA), platelet-activating factor (PAF), cytosolic phospholipase A2 (cPLA 2) , cyclooxygenase-2 (COX-2) , lipoxygenase (LOX), reactive oxygen species (ROS), prostaglandins (PGs), leukotrienes (LTs), thromboxanes (TXs), nuclear factor kappaB (NF-κB), nuclear factor κB-response element (NF-κB-RE), inhibitory subunit of NF-κB (IκB), phosphorylated IκB (IκB-P), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), inducible nitric oxide synthase (iNOS), nitric oxide (NO), superoxide (O 2 −), mitogen activated protein kinase (MAPK), and c-Jun amino-terminal kinase (JNK)
Inflammatory response also involves the recruitment of polymorphonuclear leukocytes (PMN) from the blood stream into brain tissue. The PMN migration involves chemotaxis, adhesion of PMN to endothelial cells in the area of inflammation, and diapedesis (Farooqui et al. 2007). PMN facilitate the elimination of invading antigens by phagocytosis and release free radicals and lytic enzymes into phagolysosomes. This is followed by a process called resolution, which is a turning off mechanism by neural cells to limit tissue injury. Lipoxin, another oxidized product of ARA metabolism by 5-LOX, is closely associated with resolution due to its anti-inflammatory effects. Neuro-inflammation involves several converging mechanisms responsible for sensing, transducing, amplifying, and turning off mechanisms that involve the participation of eicosanoids (Serhan et al 2007; Lawrence and Gilroy 2007). Some PGs and LTs produce proinflammatory effects while others induce antiinflammatory effects. In addition, eicosanoids also serve as autocrine factors regulating platelet aggregation, vascular tone, and edema (Farooqui 2011) . The sustained release of inflammatory mediators works to perpetuate the inflammatory cycle, activating additional microglia, promoting their proliferation, and resulting in further release of inflammatory factors. Owing to the chronic and sustained nature of the inflammation, there is often compromise of the BBB which increases infiltration of peripheral macrophages into the brain parenchyma to further perpetuate the inflammation. Collective evidence suggests that neuroinflammatory cascade is an attempt by the brain to eliminate the challenge imposed by the injury or infection, clear the system of the dead and damaged neurons, and rescue the normal functioning of this vital organ (Correale and Villa 2004). At the same time neuroinflammation promotes neurogenesis. Inflammatory factors released during mild acute neuroinflammation usually stimulate neurogenesis; whereas the factors released by uncontrolled inflammatory response block neurogenesis (Correale and Villa 2004; Helmut et al. 2011; Gomes-Leal 2012) .
Acute neuroinflammation occurs in neurotraumatic injuries, such as ischemic injury, SCI and TBI where as chronic neuroinflammation occurs in neurodegenerative diseases. Ischemic injury (stroke), a highly dynamic multifactorial metabolic insult caused by severe reduction or blockade in cerebral blood flow due to the formation of a clot. This blockade not only decreases oxygen and glucose delivery to brain tissue but also results in the breakdown of BBB and build-up of potentially toxic products in brain (Farooqui 2010). SCI and TBI are caused by mechanical trauma to spinal cord and brain. Ischemic injury, SCI and TBI trigger a complex series of biochemical and molecular mechanisms that impair the neurologic functions through the breakdown of cellular and subcellular integrity, alterations in ionic balance, increase in excitatory amino acids, elevation in intracellular calcium, activation of nitric oxide synthesis, and alterations in redox status, generation of free-radicals, induction of proinflammatory cytokine and development of neuroinflammation. Activated microglia can produce large amounts of nitric oxide, which in turn can react with superoxide to form peroxynitrite, leaving nitrotyrosine as an identifiable marker. The footprint of excess NO formation in SCI and TBI is confirmed by the increased amounts of nitrotyrosine-modified proteins (Farooqui 2010) . These mechanisms occur over a range of time, with early events within minutes of energy loss, and then progress after hours and days following the metabolic or mechanical insult leading to cell injury and tissue death (Farooqui 2010). Many of these processes are mediated by the activation of NF-κB , which is a master regulator of neuroinflammation. The activity of NF-κB is tightly regulated. Under normal conditions in the cytoplasm NF-κB occurs in bound form with its inhibitory protein, Iκ-Bα. NF-κB is activated by cytokines or neural trauma leading to the dissociation of Iκ-Bα and freeing NF-κB, which is translocated to the nucleus, where it activates the target genes containing a consensus κB site in their promoters (Chen et al. 1995). IκBα is degraded by the ubiquitin proteasome system. Mice deficient in IκBα display deregulated and sustained NF-κB activation and early postnatal lethality, highlighting a critical role of Iκ-Bα in NF-κB regulation (Beg et al. 1995). Activation of NF-κB is coupled with the stimulation of phospholipases A2, C, and D (PLA2, PLC and PLD), cyclooxygenases (COXs), lipoxygenases (LOXs), epoxygenases (EPOXs), calcium/calmodulin-dependent kinases (CaMKs), mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), nitric oxide synthases (NOS), calpains, calcinurin, and endonucleases . Activation of these enzymes results in breakdown of neural membrane phospholipids with release of arachidonic acid (ARA), which is metabolized by COX-2 and 5-LOX leading to production of proinflammatory prostaglandins (PGs), leukotrienes (LTs), and thromboxanes (TXs). The other product of PLA2-catalyzed reaction (lyso-phospholipids) is used for the synthesis of pro-inflammatory platelet-activating factor (PAF) (Fig. 2.3). While an acute insult triggers oxidative and nitrosative stress, it is typically short-lived and unlikely to be detrimental to long-term neuronal survival. Therefore, it is believed that an acute neuroinflammatory response is generally beneficial to the CNS, since it tends to minimize further injury and contributes to repair of damaged tissue (Phillis et al. 2006; Farooqui 2010, 2011).
Although, little is known about molecular mechanisms and internal and external factors that control and modulate the dynamics of acute and chronic neuroinflammation, but it is becoming increasingly evident that neuroinflammation is not only modulated by interactions among microglia, astrocytes , neurons, PMN, and endothelial cells, but also by cross-talk among various lipid mediators that originate from enzymic and non-enzymic degradation of neural membrane glycerophospholipids and sphingolipids. In addition, receptors, like TLR and transcription factors such as peroxisome proliferator-activated receptor (PPAR) also contribute to the modulation of neuroinflammation (Farooqui 2009a) .
2.3 Contribution of Astrocytes in Neuroinflammation
As stated above, astrocytes are complex, highly differentiated cells of the brain . They respond to neurotraumatic and neurodegenerative injuries by a process commonly known as reactive astrogliosis. This process involves activation of astrocytes leading to production of proinflammatory mediators, such as cytokines, chemokines, glutamate, aspartate, and quinolinic acid, reactive oxygen species (ROS) and prostanoids, (Pekny and Nilsson 2005; Correa-Cerro and Mandell 2007) (Fig. 2.3). During astrogliosis, astrocytes become hypertrophic with upregulated expression of intermediate filaments (e.g., glial fibrillary acidic protein, vimentin, nestin) and inflammatory/immune/oxidative stress markers, extracellular matrix (ECM) molecules, and growth factors and cytokines. Astrogliosis ultimately leads to the formation of glial scar as a physical barrier which inhibits axonal regeneration. Reactive astrogliosis is not merely a marker of neuropathology, but plays essential roles in orchestrating the injury response as well as in regulating the inflammation and repair in a manner that markedly impacts functional and clinical outcomes. Astrocytes contribute to neuroinflammation by interacting extensively with microglia, and can exert both pro- and anti-inflammatory effects (Farina et al. 2007) .
Astrocyte-release glutamate, which diffuses in the extrasynaptic space and may bind to glutamate receptors, including mGluRs and N-methyl-D-aspartate receptors (NMDARs) on neighboring presynaptic terminals, activating PLC-mediated signaling and modulating the release of neurotransmitter (Jourdain et al. 2007; Bonansco et al. 2011). Formation of astrocytic scar not only acts as neuroprotective barriers to inflammatory cells and infectious agents, but is also promotes reorganization and induces structural changes that are long lasting and persist long after the triggering insult may have resolved (Sofroniew and Vinters 2010). The migration of astrocytes is a critical step in the formation of a densely-packed glial scar (Saadoun et al. 2005) and TGF-β1 has been reported to play an important role in glial scar formation (Kohta et al. 2009). Glial scar formation is prevented by cysteinyl leukotrienes (CysLT) receptor antagonists or 5-LOX inhibitors supporting the view that cysteinyl leukotriene receptor 1 (CysLT1R) are closely associated with astrocyte proliferation and glial scar formation after brain injury (Yu et al. 2005; Zhou et al. 2006) . Astrocytes not only participate in the local innate immune responses through the involvement of TLRs, nucleotide-binding oligomerization domains, double-stranded RNA-dependent protein kinase, scavenger receptors, mannose receptor and components of the complement system, but also play an important role in the neuroinflammation and tissue repair through the secretion of soluble mediators, such as CXCL10, CCL2, interleukin-6 and BAFF (Zhao et al. 2011). In brain TLRs are not only involved in peripheral innate immunity but may also play a role in the development and regulation of CNS inflammation, neurodegeneration and brain trauma. These receptors initiate downstream signaling to activate the key transcription factor, NF-κB, producing inflammatory cytokines (Kawai and Akira 2010; Crack and Bray 2007). Recent studies have indicated that in primary cultures of human astroglial (HAG) cells miRNA-146a is an important regulator of the innate immune response and pro-inflammatory signaling (Aronica et al. 2010) . It is shown that miRNA-146a modulates the interleukin-1 receptor-associated kinase-1 and 2 (IRAK-1 and IRAK-2) expression in IL-1β + Aβ42-treated HAG cells . IRAK-1 and IRAK-2 are essential components of Toll-like/IL-1 receptor signaling. Using miRNA-146a-, IRAK-1-, or IRAK-2 promoter-luciferase reporter constructs, it is shown that the decrease in IRAK-1 and increase in miRNA-146a and IRAK-2 expression in interleukin-1β (IL-1β) and amyloid-β-42 (Aβ42) peptide-stressed HAG cells are closely associated with neuroinflammation. MyD88, a mammalian adapter protein serves as a bridge between TLR4 and IL-1 receptor associated kinase (IRAK1) that then recruits into the complex TNF receptor-associated factor 6 (TRAF6) (Aronica et al. 2010; Saugstad 2010) . This chain of events triggers the activation of I-κB kinase and JNK, which in turn, modulates the downstream NF-κB, a transcription factor located in the cytoplasm. The dissociation of NF-κB-I-κB complex results in translocation of free NF-κB to the nucleus , where it binds to target sequences in the genome and facilitates the expression of a number of proteins including many enzymes (sPLA2, COX-2, NADPH oxidase, and inducible nitric oxide synthase (iNOS) , NADPH oxidase and matrix metalloproteinases (MMPs)) and cytokines (TNF-α, IL-1β, and IL-6) , chemokines, and other proteins including, intracellular adhesion molecule-1 (ICAM-1), vascular adhesion molecule-1 (VCAM-1), E-selectin (Table 2.1). The production of above inflammatory mediators is regulated by the negative feedback provided by the hypothalamus-pituitary-adrenal (HPA) axis (Farooqui 2010) .
Table 2.1
Cytokines, chemokines, and other inflammatory metabolites associated with the development of neuroinflammation
Cytokines/chemokines/factors | Nature | Effect | Reference |
|---|---|---|---|
TNF-α | Proinflammatory | Increased | Block and Hong 2005 |
IL-1β | Proinflammatory | Increased | Block and Hong 2005 |
IL-6 | Proinflammatory | Increased | Block and Hong 2005 |
IL-17 | Proinflammatory | Increased | Block and Hong 2005 |
IL-15 | Proinflammatory | Increased | Block and Hong 2005 |
IL-18 | Proinflammatory | Increased | Block and Hong 2005 |
IL-4 | Antiinflammatory | Increased | Block and Hong 2005 |
IL-10 | Antiinflammatory | Increased | Block and Hong 2005 |
IL-13 | Antiinflammatory | Increased | Block and Hong 2005 |
TGF-α | Proinflammatory | Increased | Block and Hong 2005 |
Chemokine MIP-1α | Proinflammatory | Increased | Block and Hong 2005 |
Chemokine MCP-1 | Proinflammatory | Increased | Block and Hong 2005 |
Chemokine MDC | Proinflammatory | Increased | Block and Hong 2005 |
ICAM-1 | Proinflammatory | Increased | Block and Hong 2005 |
VCAM-1 | Proinflammatory | Increased | Block and Hong 2005 |
E-selectin | – | Increased | Block and Hong 2005 |
A second group of transcription factors called PPARs has also been implicated in the neuroinflammation (Drew et al. 2005). PPARs are expressed in several cell types of the brain including microglia, astrocytes, oligodendrocytes and neurons (Heneka and Landreth 2007). They regulate gene expression using various ligand-dependent and—independent molecular processes. Three different isoforms of the PPARs exist and they are encoded by separate genes: PPAR- γ, PPAR-α, and PPAR-δ (Torra et al. 2001). They not only act as nutritional sensors, but also regulate inflammation, cellular growth, differentiation, and apoptosis. Activation of PPAR isoforms elicits anti-inflammatory activities in neural cells. Lysophosphatidic acid, nitrolinoleic acid, 9- and 13-hydroxyoctadecadienoic acids (9- and 13-HODE), 15-hydroxyeicosatetraenoic acid (15-HETE)), some eicosanoids (prostaglandin D2 (PGD2), and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2)), and oxidized phospholipids are endogenous ligands for PPAR-γ. Prior to ligand binding, however, PPARs heterodimerize with retinoid X receptor (RXR), forming a complex . This complex is required for binding to specific DNA sequences, known as PPAR response elements, in the promoter region of target genes (Fig. 2.4). Upon binding to their ligands, PPARs undergo conformational changes allowing the release of co- repressors, and recruitment of coactivators, followed by the activation of transcription (Farooqui et al. 2004; Feige et al. 2006). Mice deficient in PPAR have a prolonged response to inflammatory stimuli. PPAR ligands, in particular those of PPARα and PPARγ, inhibit the activation of inflammatory gene expression and can negatively interfere with pro-inflammatory transcription factor signaling pathways in vascular and inflammatory cells (Moraes et al. 2006). It is becoming increasingly evident that activities of a number of transcription factors, for example NF-κB, AP-1 (activator protein-1), and STAT-1 (signal transducer and activator of transcription) are inhibited by PPARs either via direct interaction or by competition for limited supplies of coactivators (Daynes and Jones 2002; van Neerven and Mey 2007). In addition, PPARγ activation in microglial cells not only inhibits the expression of inflammatory cytokine and iNOS, and decrease in NO production, but also suppresses the expression of COX-2 and the generation of PGs (Combs et al. 2000) (Fig. 2.4) . PPARγ agonists have also been demonstrated to suppress the Aβ-mediated activation of microglia in vitro and prevente cortical or hippocampal neuronal cell death (Combs et al. 2000; Kim et al. 2002; Luna-Medina et al. 2005). These functions are important in regard to the anti-inflammatory properties of PPARs since proinflammatory gene expression. While PPAR-γ directly regulates inflammatory gene expression, it also interferes with the activation of NFκB creating an intriguing interaction between these two transcription factors (Van den Berghe et al. 2003; Heneka et al. 2005). Both NFκB and PPAR-γ may be regulated by n-3 PUFAs. In addition, retinoic acid (RA) is an active metabolite of retinoid metabolism. It regulates a wide range of biologic processes, including cell proliferation, differentiation, and morphogenesis (Farooqui et al. 2004). Recent studies suggest that in microglial cells inhibition of NF-kB translocation to the nucleus and suppression of JAK/STAT pathways may contribute to the anti-inflammatory mechanisms of RA (Dheen et al. 2005). Since neuroinflammation is a risk factor of neurodegenerative diseases, the anti-inflammatory effect of RA may provide a novel therapeutic value for the treatment of neurodegenerative diseases, AD, PD, and ischemic injury (Farooqui 2009a) .
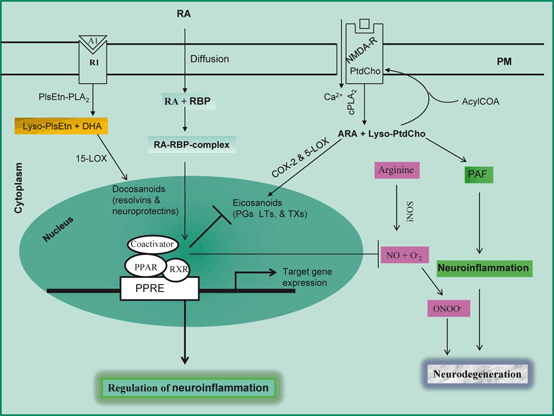
Fig. 2.4
Modulation of neuroinflammation by PPAR. Phosphatidylcholine (PtdCho), lyso-phosphatidylcholine (lyso-PtdCho), arachidonic acid (ARA), platelet-activating factor (PAF), cytosolic phospholipase A2 (cPLA 2), cyclooxygenase-2 (COX-2) , reactive oxygen species (ROS), 5-lipoxygenase (5-LOX), 15-lipoxygenase (15-LOX), Ethanolamine plasmalogen (PlsEtn), plasmalogen-selective phospholipase A2 (PlsEtn-PLA 2) , Lyso-plasmalogen (Lyso-PlsEtn), prostaglandins (PGs), leukotrienes (LTs), retinoic acid (RA), retinoic acid binding protein (RBP), peroxisome proliferator-activated receptor (PPAR), PPRA response element (PPRE), retinoid X receptor (RXR), inducible nitric oxide synthase (iNOS); and superoxide (O 2 −)
2.4 Differences Between Acute and Chronic Neuroinflammation
Acute neuroinflammation develops rapidly with the experience of pain, whereas chronic inflammation develops slowly. Acute neuroinflammation is accompanied by rapid activation of microglia and astrocytes, damage to the BBB , and acute up-regulation of proinflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF-α) and IL-6 (Schmidt et al. 2005; Farooqui 2010). The resolution of acute inflammation is complex and involves several distinct cellular mechanisms. Cell clearance is critical to resolution and is driven both by apoptosis of leukocytes (Rossi et al. 2006) and egress from tissues (Uller et al. 2006). Clearance of the inflammatory site is mediated in part via the non-phlogistic recruitment of monocytes that, as macrophages, participate in the phagocytosis of apoptotic cells (Schwab et al. 2007). Thus, in non-neural tissues the resolution of inflammation is a dynamic process, which is orchestrated by lipid mediators that play important counter-regulatory roles including cytokines, chemokines, and lipid mediators such as lipoxins, resolvins, and protectins (Serhan 2009; Farooqui 2011). These mediators decrease vascular permeability and retard PMN recruitment, while promoting recruitment of monocytes and stimulating efferocytosis (Serhan et al. 2008). In the brain, the acute inflammatory response also involves the recruitment of neutrophils and monocytes to the site of injury in an attempt to aid the injured tissue via the secretion of above mentioned cytokines and other signaling molecules. In addition to the recruitment of neutrophils and monocytes, the brain also possesses native cells capable of organizing and expressing glial fibrillary acidic protein (GFAP) (Szmydynger-Chodobska et al. 2012; Schiff et al. 2012).
Chronic neuroinflammation differs from acute inflammation in that it is below the threshold of pain perception. As a result, the immune system continues to attack at the cellular level. Chronic inflammation lingers for years causing continued insult to the brain tissue reaching the threshold of detection (Wood 1998) and initiating pathogenesis of chronic disease. Chronic inflammation disrupts hormonal signaling networks not only in the brain, but also in the visceral organs. As stated earlier, chronic neuroinflammation is accompanied by long-standing activation of microglia and subsequent sustained release of inflammatory mediators leading to increase in oxidative and nitrosative stress (Tansey et al. 2007). The sustained release of inflammatory mediators not only alters the inflammatory cycle and activates additional microglial cells , but also promotes proliferation, leading to further release of inflammatory factors. In non-neural tissues sustained inflammation due to persistent activation of inflammatory cells or defects in the resolution program results in a fibrogenic response, which involves the overall remodeling of tissue structure and eventual organ failure. Fibrogenic response is supported not only by sustained inflammation and cell proliferation, but also by growth factor responses. Owing to the chronic and sustained nature of the inflammation, there is often compromise of the BBB which increases infiltration of peripheral macrophages into the brain parenchyma to further perpetuate the inflammation (Rivest 2009). Rather than serving a protective role as does acute neuroinflammation, chronic neuroinflammation is most often detrimental and damaging to the brain tissue. Thus, whether neuro-inflammation has beneficial or harmful outcomes in the brain may depend critically on the duration of the inflammatory response. Prolonged chronic inflammatory state has detrimental health effects and predisposes to a wide variety of chronic diseases, especially those that are more prevalent with advanced age, such as cardiovascular diseases, diabetes, and neurodegenerative diseases (Farooqui et al. 2007). Chronic inflammation is also a strong predictor of both disability and mortality in the elderly—even in the absence of clinical disease (Penninx et al. 2004; Farooqui 2013). Although, significant information is available on the generation of proinflammatory mediators, but little is known about internal and external factors that modulate the dynamic aspects of acute and chronic neuroinflammation. Depending on its timing and magnitude in brain tissue, neuroinflammation serves multiple purposes. It is involved in protection of uninjured neurons and removal of degenerating neuronal debris and also in assisting repair and recovery processes (Farooqui 2010, 2011). It is not yet known whether neuroinflammatory events precede disease states or are a direct consequence of the damage that occurs in pathology of neurodegenerative diseases. For example, Aβ plaques have been shown to induce proinflammatory effects in animal models of AD (Halliday et al. 2000; Tuppo and Arias 2005) supporting the view that neuroinflammatory events initiate or even aid in the progression of AD (Heneka and O’Banion 2007; Bales et al. 2000).
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


