Fig. 9.1
Oxidative stress-mediated modulation of transcription factors leads to gene transcription involved in neural cells survival and death. Glutamate (Glu), N-Methyl-D-aspartate receptor (NMDA-R), Phosphatidylcholine (PtdCho), cytosolic phospholipase A2 (cPLA2), Reactive oxygen species (ROS), reactive nitrogen species (RNS), 4-hydroxynonenal (4-HNE), Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), nuclear factor-κB (NF-κB), Forkhead box O (FoxO), arginine (Arg), c-Jun NH(2)-terminal kinase (JNK), nitric oxide (NO), Peroxynitrite (ONOO-), Activator protein 1 (AP1), Jun amino-terminal kinases (JNK), Hypoxia-inducible factor 1-alpha (HIF-1α), and Forkhead family of transcription factors (FOXO)
Collectively, these studies suggest that signal transduction mechanisms involved in neuronal survival or death are closely related and interconnected. In fact, some shared mechanisms between neural cell survival and neurodegeneration have been reported (Farooqui 2012). Thus, tumor suppressor gene p53 is activated by ROS in a dose-dependent manner. In response to the activation of p53, other pathways are induced, which involve mitogen-activated protein kinases (MAPKs) like JNK and p38 (Klein and Ackerman 2003). However, the overall response of neurons to oxidative stress depends on which pathways are predominantly stimulated. For example, induction of the MAPKs (JNK and p38) is accompanied by activation of heme oxygenase-1 (HO-1), which counteracts the effects of ROS (Aggeli et al. 2006). Moreover, induction of cyclooxygenase-2 (COX-2) ameliorates the harmful effects of ROS through its negative effects on p53 (Han et al. 2002). These complex interactions between pro-apoptotic and pro-survival pathways suggest that the neurons turn on genes and induce pathways to prevent cell death and eventually leading to morphological and functional defects on the brain except in extreme conditions when they resort to apoptosis (Fig. 9.2) (Farooqui 2012).
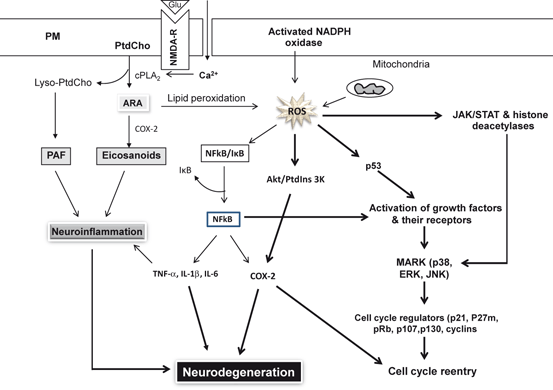
Fig. 9.2
Interactions between pro-apoptotic and pro-survival pathways. Glutamate (Glu), N-Methyl-D-aspartate receptor (NMDA-R), Phosphatidylcholine (PtdCho), lysophosphatidylcholine (lyso-PtdCho), arachidonic acid (ARA), cytosolic phospholipase A2 (cPLA 2 ), cyclooxygenase-2 (COX-2), platelet activating factor (PAF), Reactive oxygen species (ROS), nuclear factor-κB (NF-κB), inhibitory subunit of NF-κB (I-κB), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), protein 53 (p53), mitogen-activated protein kinase (p38), MAPK (Erk), and c-Jun NH(2)-terminal kinase (JNK)
9.2.1 Genes Driven by Transcription Factor, Nrf2
NRF2 (nuclear factor (erythroid-derived 2)-like 2) is a redox sensitive transcriptional factor, which is present in the cytoplasmic compartment in an inactive form (complexed with Kelch-like ECH-associated protein 1, Keap1) (Fourquet et al. 2010). At steady state, Keap1 acts as an adaptor for an E3 ubiquitin ligase, thereby targeting Nrf2 to the proteasome for degradation (Kobayashi et al. 2004).
In response to mild oxidative stress (low ROS, such as H2O2) and mild nitrosative stress (low NO), NRF2 dissociates from Keap1 due to oxidation of its several cysteine resisues. Free NRF2 (Nrf2) migrates to the nucleus and dimerizes with other basic-leucine zipper (bZIP) proteins such as small Maf proteins to form a transactivation complex that binds to antioxidant response elements (AREs), which are cis-acting regulatory elements identified within the 59-flanking regions of such phase II detoxification enzymes (Fig. 9.3) (Rushmore et al. 1991; Dhakshinamoorthy and Porter 2004; Motohashi and Yamamoto 2004). Antioxidant enzyme systems regulated by Nrf2 include, but not limited to, redox regulation [superoxide dismutase (SOD), catalase (CAT), sulfaredoxin (Srx), thioredoxin (Trx), peroxiredoxin (Prdx) system], glutathione synthesis and metabolism [glutathione peroxidase (Gpx), glutathione reductase (GR), γ-glutamine cysteine ligase (GCL) and γ-glutamine cysteine synthase (GCS)], quinone recycling [NAD(P)H quinone oxidoreducase (NQO1)] and iron homeostasis [heme oxygenase 1 (HO-1), Ferritin] (Itoh et al. 1997; Chen and Kunsch 2004). These enzymes increase neural cell survival by protecting against oxidative stress. This regulated adaptive response is called as the “Phase II detoxification response” (Fig. 9.3) (Prestera et al. 1993).
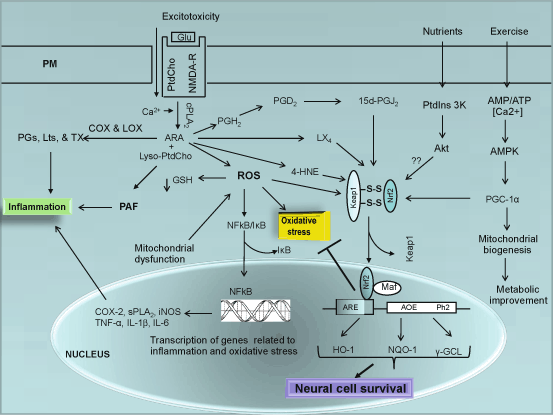
Fig. 9.3
NF-κB and Nrf2 signaling in the brain. Glutamate (Glu), N-Methyl-D-aspartate receptor (NMDA-R), Phosphatidylcholine (PtdCho), lysophosphatidylcholine (lyso-PtdCho), cytosolic phospholipase A2 (cPLA 2 ), cyclooxygenase-2 (COX-2), lipoxygenase (LOX), arachidonic acid (ARA), platelet activating factor (PAF), Reactive oxygen species (ROS), nuclear factor-κB (NF-κB), inhibitory subunit of NF-κB (I-κB), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), 4-hydroxynonenal (4-HNE), prostaglandin H2 (PGH 2 ), prostaglandin D2 (PGD 2 ), prostaglandin J2 (PGJ 2 ), peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC – 1), Heme oxygenase (HO-1), NADPH quinine oxidoreductase (NQO-1), and γ-glutamate cysteine ligase (γ-GCL)
In addition to mild oxidative stress, other lipid mediators (4-HNE, 15-dPGJ2, and lipoxin 4 (LX4)) also promote the dissociation of Nrf2 from Keap1 complex and facilitate the migration of Nrf2 from cytosol to the nucleus. All these lipid mediators are derived from arachidonic acid (ARA). LXA4, an endogenously produced eicosanoid, which inhibits neutrophil recruitment and activation, reduces many cell responses evoked by pro-inflammatory cytokines, promotes resolution of inflammation, and acts as an endogenous “braking signal” in the inflammatory process (Serhan and Chiang 2008; Hecht et al. 2009), upregulates the expressions of HO-1 mRNA and protein via activation of p38 MAPK pathway, nuclear translocation of Nrf2 and Nrf2 binding to the HO-1 ARE, but not via activation of PtdIns 3-K/Akt or ERK pathway (Chen et al. 2013) supporting the view that eicosanoids may also contribute to phase II detoxification.
Nrf2-deficient mice are highly susceptible to chemical-induced toxicity, carcinogenesis and oxidative burden (Kensler et al. 2007). In addition, other gene classes including those associated with protein transport, ubiquitination, phosphorylation, cell cycle, growth and apoptosis have been identified as potentially Nrf2-dependent and are altered in studies with phenolic antioxidant and isothiocyanate treatments (Barve et al. 2008; Wakabayashi et al. 2010). Accumulating evidence suggests that the roles of Nrf2 modulated genes are to (a) provide direct antioxidants, (b) encode enzymes that directly inactivate oxidants, (c) increase levels of glutathione synthesis and regeneration, (d) stimulate NADPH synthesis, (e) enhance toxin export through the multidrug-response transporters, (f) enhance the recognition, repair, and removal of damaged proteins, (g) elevate nucleotide excision repair, (h) regulate expression of other transcription factors, growth factors and receptors, and molecular chaperones, and (i) inhibit cytokine-mediated inflammation (Hayes and McMahon 2009; Kensler et al. 2007). Above mentioned processes are supported by the transcription of more than 200 genes involved in the cellular antioxidant and anti-inflammatory defense (Kensler et al. 2007; Wakabayashi et al. 2010). It is well known that post-translational modifications such as phosphorylation play a major role in the regulation of gene expression and function. These covalent modifications are closely associated with the control of intracellular distribution, transcriptional activity, and stability of Nrf2 (Surh et al. 2008). Some transcription factors antagonize the effect of Nrf2 either through the binding with AREs or by inhibiting Nrf2 through a physical association. Thus, small MAF proteins, BACH1, and the immediate early proteins c-FOS and FRA1 can compete with Nrf2 for binding to AREs (Nguyen et al. 2000). In addition, some transcription factors (activating transcription factor 3, proliferator-activated receptor γ (PPAR)γ, and retinoic acid receptor α) form complex with NRf2 and inhibit its activity. (Brown et al. 2008; Ikeda et al. 2000; Wang et al. 2007), whereas other receptors, transcription factors, and contain pathways (arylhydrocarbon receptor (AhR), NF-κB, p53, and Notch pathways) modulate Nrf2 activity through transcriptional cross-talk (interplay) (Wakabayashi et al. 2010).
9.2.2 Genes Driven by Transcription Factor, Ap-1
The AP-1 transcription factor is composed of Jun-Jun homodimers or heterodimers of members of the Jun (c-Jun, JunB and JunD) and Fos (c-Fos, FosB, Fra-1 and Fra-2) families (Angel and Karin 1991; Morgan and Curran 1995). Neurotransmitters, growth factors, cytokines, mild oxidative stress, and oncoproteins stimulate AP-1 activity (Fig. 9.4). AP-1 plays an important role in the regulation of neural gene expression through extracellular signals. AP-1 is composed of multiple proteins, which bind as heterodimers to the DNA sequence TGACTCA. Two regulatory mechanisms are involved in the activation of AP-1 (Morgan and Curran 1995). In the first mechanism, some AP-1 proteins (such as c-Jun) are encoded by immediate early genes that are transcriptionally induced. c-fos and c-jun are also induced by low levels of 4-hydroxynonenal (4-HNE), a metabolite, which is derived from nonenzymic oxidation of arachidonic acid during oxidative stress. In addition, hydroperoxy fatty acids and H2O2 promote the expression of c-fos and Jun 2 proteins that form heterodimers, which activate AP-1 (Rao et al. 1995; Shaulian and Karin 2002) . Activation of AP1 through this mechanism is involved in genes regulating cell cycle progression leading to neural cell survival. Thus, activation of early response genes is involved in the transition from the GI into the S phase of the cell cycle (Nestler and Hayman 2002). Upregulation of c-fos and c-jun is accompanied by increase in mRNA, protein, or AP-1 DNA binding activity. They are markers of the immediate early gene response that plays a key role in the control of terminal cell differentiation and proliferation in a number of cell types (Nestler and Hayman 2002).
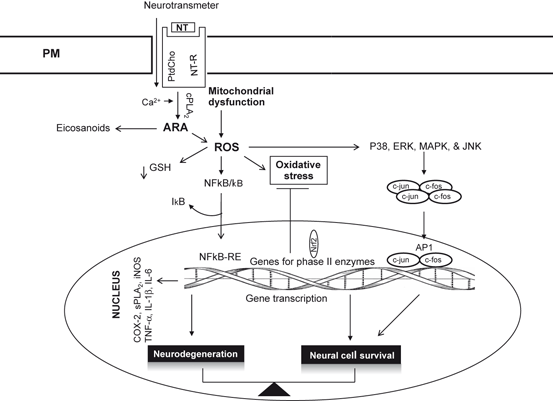
Fig. 9.4
AP1-mediated gene expressiong in the brain. Neurotransmitter (NT), neurotransmitter receptor (NT-R), phosphatidylcholine (PtdCho), cytosolic phospholipase A2 (cPLA 2 ), arachidonic acid (ARA), reactive oxygen species (ROS), glutathione (GSH), nuclear factor-κB (NF-κB), inhibitory subunit of NF-κB (I-κB), tumor necrosis factor-α (TNF-α), interleukin-1β(IL-1β), interleukin-6 (IL-6), proto-oncogene c-Fos (c-fos), proto-oncogene c-jun (c-jun), activator protein-1 (AP-1), mitogen-activated protein kinase (p38), MAPK (Erk), and c-Jun NH(2)-terminal kinase (JNK)
In the second mechanism, AP-1 activity is regulated by posttranslational modifications, such as phosphorylation by mitogen-activated protein kinases (MAPKs), which comprises the extracellular signal-regulated kinase, p38 MAPK, and c-Jun NH2-terminal kinase. The exact mechanism of a specific condition or treatment on AP-1 activation and the relative role of different MAPKs in these processes are diverse. Upon activation, AP-1 interacts with the 12-O-tetradecanoylphorbol-13-acetate response element and induces transcription of a variety of genes involved in multiple cellular functions, such as proliferation, survival, differentiation, and transformation.
Many genes expressed in the brain contain AP-1 sites within their regulatory regions (Morgan and Curran 1995; Shaulian and Karin 2002). For examples AP-1 binding site is present in genes encoding neuropeptides (neurotensin and substance P), neurotransmitter receptors (D1 dopamine, NR1 NMDA, and GluR2 AMPA glutamate receptor subunits), neurotransmitter synthetic enzymes (tyrosine hydroxylase), and cytoskeletal proteins (neurofilament proteins). The antioxidant responsive element (ARE) in the promoter region of the human NQO1 gene also contains AP-1 or AP-1-like DNA binding sites, AP-1 proteins have been implicated in the formation or function of this and other ARE complexes (Li and Jaiswal 1992), which are sensitive to the changes in cellular redox states associated with hypoxia and hypoxia-reoxygenation (Ausserer et al. 1994). It should be noted that AP-1 dimers are part of the transcriptional effector system of the ERK, JNK and p38 MAP kinases supporting the view that AP1 proteins may be associated with gene transcription as well as a wide range of action by post-translational modification of non-nuclear substrates. Accumulating evidence suggests that AP-1 proteins in the adult brain may be associated with neuroprotection and neurodegeneration (Herdegen and Waetzig 2001).
9.2.3 Genes Driven by Transcription Factor NF-κB
NF-κB (nuclear factor-κB) proteins are a family of transcription factors, which are involved in neuroinflammation, immune response, apoptosis, development, and cell growth (Stephenson et al. 2000; Imielski et al. 2012) . NF-κB is composed of five DNA binding proteins sharing the N-terminal Rel-homology domain (RHD): NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), cRel, and RelB that recognize a common sequence motif. NF-κB is found in neuronal and glial cells, and is involved in activation and modulation of a large number of genes in response to oxidative stress. Five different proteins of NF-κB factor, namely p50, RelA/p65, c-Rel, RelB, and p52, can combine differently to form active dimers in response to external stimuli. RelA is activated by neurotoxic agents while c-Rel produces neuroprotective effects (Sarnico et al. 2009; Imielski et al. 2012). RelA, RelB, and c-Rel share a transactivation domain (TAD) located in the C-terminus, while the NF-κB1 (p50) and NF-κB2 (p52) are processed from larger precursors (p105 and p100) respectively that do not possess a TAD (Nabel and Verma 1993). Homodimers of p50/p50 or p52/p52 inhibit gene transcription, while dimers composed of at least one TAD-positive subunit positively regulate gene expression. NF-κB is activated by mechanisms that disrupt its binding with IκB, resulting in translocation of the liberated NF-κB to the nucleus where it binds to κB elements in the promoter and enhancer regions of responsive genes, leading to gene induction or gene repression (Perkins 2006). Several other IκB-like ankyrin-repeat containing NF-κB binding proteins have been reported to modulate nuclear NF-κB transcriptional activity on a subset of genes (Yamamoto et al. 2004) or, when over-expressed, prevent nuclear localization of NF-κB (Naumann et al. 1993) . The canonical activation mechanism involves the phosphorylation of IκBα on serines 32 and 36, which marks IκBα for ubiquitylation and proteasome-mediated degradation. Another activation mechanism (noncanonical) involves the phosphorylation of IκBα on tyrosine 42 (Tyr42), which results in the dissociation of IκBα from NF-κB without proteasome-dependent degradation (Imbert et al. 1996). Recent studies have indicated the occurrence of a distinct group of atypical IκB proteins. These proteins are called as the BCL-3 subfamily. These atypical IκBs show entirely different sub-cellular localizations, activation kinetics and an unexpected functional diversity (Schuster et al. 2013). First of all, interaction of BCL-3 with NF-κB transcription factors occurs in the nucleus in contrast to classical I-κBs, whose binding to NF-κB predominantly take place in the cytoplasm. Secondly, BCL-3 is strongly induced after NF-κB activation not only by lipopolysaccharide (LPS), but also by IL-1β-mediated stimulation of B cell and T cell antigen receptors (Schuster et al. 2013). Unlike I-κB, BCL-3 is not degraded through proteasome-dependent degradation. Finally, the interaction of BCL-3 with DNA-associated NF-κB transcription factors can further enhance or diminish their transcriptional activity. Thus, they do not exclusively act as inhibitors of NF-κB activity. The capacity to modulate NF-κB transcription either positively or negatively, represents their most important and unique mechanistic difference to classical IκBs. Several reports have revealed the importance of atypical IκB proteins for immune homeostasis and the severe consequences following their loss of function (Schuster et al. 2013) .
During oxidative stress, RelA and p50 factors are rapidly activated, but how they associate with c-Rel to form active dimers and contribute to the changes in the diverse dimer activation for neuron susceptibility is unknown. Oxidative stress produces persistently activation of RelA and p50 factors of NF-κB in neurons that are destined to die. There are several potential routes through which NF-κB can act to induce neuronal death, including induction of death proteins and an aborted attempt to reenter the cell cycle. Under normal conditions, p50 and p65 protein subunits of NF-κB reside in the cytoplasm as an inactive complex bound by inhibitor proteins, I-κBα and I-κBβ. In response to oxidative stress, I-κB is phosphorylated by I-κB kinase. I-κB is ubiquitinated and degraded by the proteasome; simultaneously, the active heterodimer translocates to the nucleus where it initiates gene transcription targeting binding sites homologous to the canonical DNA sequence 5′-GGGACTTTCC-3′ in the regulatory regions of NF-κ B-sensitive genes (Meffert and Baltimore 2005; Wong et al. 2011) . The mechanism by which NF-κB mediates cell death remains unknown. It is proposed that the migration of NF-κB from cytoplasm to the nucleus results in its binding with target sequences in the genome and facilitates the expression of a number of proteins including many enzymes (sPLA2, COX-2, NADPH oxidase and inducible nitric oxide synthase, superoxide dismutase) and proinflammatory cytokines (TNF-α, IL-1β, and IL-6) . Activation of p50/RelA complex in the nucleus also induces the pro-apoptotic Bim and Noxa genes. Upregulation of sPLA2, COX-2, NADPH oxidase and inducible nitric oxide synthase, and cytokines is closely associated with neuronal cell death following severe oxidative stress-mediated neuronal injury .
NF-κB also plays an important role in neuronal survival. Although the molecular mechanism of NF-κB-mediated neuroprotection is not fully understood. However, recent studies have indicated that c-Rel-containing dimers, p50/c-Rel and RelA/c-Rel, but not p50/RelA, promote Bcl-xL transcription (Sarnico et al. 2009). Thus, the oxygen glucose deprivation (OGD) of cortical neurons not only results in Bim induction but also downregulation of Bcl-xL promoter activity and reduction in endogenous Bcl-xL protein content. These findings indicate that within the same neuronal cell, the balance between activation of p50/RelA and c-Rel-containing complexes fine-tunes the threshold of neuron vulnerability to the ischemic insult (Sarnico et al. 2009). NF-κB dimer (p50/p65) participates in the pathogenesis of postischemic injury by inducing pro-apoptotic gene expression, while c-Rel-containing dimers increase neuron resistance to ischemia by inducing anti-apoptotic gene transcription (Pizzi et al. 2009). In addition, NF-κB activation may prevent neuronal cell death through the induction of inhibitor of apoptosis proteins (IAPs) and manganese superoxide dismutase (Mn-SOD). NF-κB-mediated neuroprotective signaling produces changes in the structure and function of neuronal circuits (Mattson and Meffert 2006). Collective evidence suggests that constitutive NF-κ B signaling is critically homeostatic to many aspects of normal brain function, and essential to the regulation of cell proliferation, apoptosis, innate, and adaptive immunity, the inflammatory response, synaptic plasticity, neurite growth, formation of functional dendritic spines, and related stress responses. NF-κ B activation is also an important part of a cellular recovery process that may protect neural cells against oxidative-stress or brain trauma-induced apoptosis and neurodegeneration (Mattson and Meffert 2006; Kaltschmidt and Kaltschmidt 2009; Gavalda et al. 2009; Boersma et al. 2011) suggesting that the ultimate survival or death of neurons depends on which, where, and when the NF-κB factors are activated .
The Nrf2 and NF-κB signaling pathways interact with each other at several points to control the transcription or function of downstream target proteins and gene transcription. Thus, Nrf2 and NF-κB compete with each other for ARE site on DNA and NF-κB has been recently shown to retard the transcription of Nrf2-dependent genes by reducing available co-activator levels and promoting recruitment of a co-repressor. In addition, p65 and Nrf2 both bind to the CH1-KIX domain of CREB-binding protein (CBP), and after phosphorylation of p65 at Ser276, NF-κB suppresses transcription of ARE-dependent genes by preventing CBP from binding to Nrf2 (Liu et al. 2008). A second mechanism of p65 transcriptional repression of the ARE involving HDAC3 has also been described. Overexpression of p65 causes the recruitment of HDAC3 to the ARE by binding to CBP or MafK. HDAC3 was shown to bind MafK in the NRF2 dimerization region and to prevent the acetylation of MafK by CBP (Liu et al. 2008). As stated above, targets of NF-κB include genes for proinflammatory cytokines, chemokines , immunoreceptors, cell-adhesion molecules, stress-response genes, regulators of apoptosis, growth factors, and transcription factors, among many others .
9.2.4 Genes Driven by Transcription Factor, FOXO
The Forkhead box class O (FOXO) subfamily of transcription factors includes at least four members (FOXO1, FOXO3a, FOXO4, and FOXO6). These factors play important roles in metabolism, organ development, cell cycle, apoptosis, DNA repair and oxidative stress resistance in neural and non-neural cells (Burgering 2008; Huang and Tindall 2007). FOXO family members act as either transcriptional activators or repressors by forming complexes with different transcriptional modulators. Their function is tightly regulated by the upstream phosphoinositide 3-kinase (PtdIns 3K) and Akt (PKB) pathway, which in turn induces the phosphorylation of FOXO factors and their nuclear export into the cytoplasm (Brunet et al. 1999) inhibiting the FOXO-stimulated transcription of target genes. FoxO family members activate transcription by specifically binding to apparently shared binding sites (the consensus sequence is GTAAACA) in the promoters of target genes (Pierrou et al. 1994). Studies on FOXO-mediated oxidative stress resistance have indicated that FOXO1 or FOXO3 expression leads not only to decrease in neural cell death, decrease in production of ROS, and increase in expression of antioxidants, autophagy-related genes, and antiapoptotic proteins (Burgering 2008). In neural and non-neural cells, oxidative stress mediated activation of FOXOs id linked with modulation of 5-AMP-activated protein kinase (AMPK) and sirtuins (class III histone deacetylases, Sirt1 and Sirt3) (Greer et al. 2007; Sundaresan et al. 2008). As stated in chapters 1 and 6, AMPK is an important sensor and regulator of cellular energy status that responds to energy depletion by stimulating ATP production (Li et al. 2009). In cultured fibroblasts, AMPK-mediated activation of FoxOs leads to increase in expression of several genes that are important for controlling energy balance and oxidative stress resistance (Greer et al. 2007). Additionally, AMPK not only regulates endothelial function (Zou et al. 2002), angiogenesis (Nagata et al. 2003), and the cell cycle (Guo et al. 2007), but also inhibits vascular inflammation (Gaskin et al. 2007), and prevents endothelial injury induced by hyperglycemia and FFAs (Ido et al. 2002). Likewise, SIRT1 and SIRT3 (NAD + -dependent protein/histone deacetylase), which are antiaging and antiinflammatory proteins, are activated upon induction of oxidative stress and are protective from oxidative damage through induction of antioxidants SOD2 and catalase in the heart (Sundaresan et al. 2008; Rajendran et al. 2011). Taken together, it is increasingly recognized that FOXOs are necessary for induction of antioxidant gene expression and protection against oxidative injury is consistent with their being critical transcriptional mediators of AMPK and Sirt-mediated neuroprotection .
9.2.5 Genes Deriven by Hypoxia Inducible Factor-1
The transcription factor hypoxia-inducible factor-1 (HIF-1) is a heterodimer consisting of an oxygen-sensitive HIF-1α subunit and a constitutively expressed HIF-1β subunit. HIF-1α possesses an oxygen-dependent degradation domain containing two key proline residues which are hydroxylated by HIF prolyl-hydroxylases (PHDs) in the presence of oxygen (Kaelin and Ratcliffe 2008; Landriscina et al. 2009). This transcription factor regulates cellular adaptation and survival under hypoxic stress. HIF-1 binds to hypoxia-response elements (HRE) found in promoter or enhancer DNA regions of target hypoxia-inducible genes that include vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT-1), nitric oxide synthases, along with between 100 and 200 other genes (Kaelin and Ratcliffe 2008). Activation of HIF-1 is involved in many physiological and pathological processes including, vascular remodeling, neuroinflammation, and hypoxia/ischemia-mediated brain damage. Many studies have indicated ROS induced alterations in HIF-1α activity but information about the exact kinetics and conditions of ROS production and their specific relevance to HIF-l α remains unknown. Recent studies have indicated that high levels of ROS lead to the stabilization of hypoxia inducible factors (HIFs) by inhibiting the activity of prolyl hydroxylases (PHDs), which are involved in the oxygen-dependent destabilization of HIF proteins (Kaelin and Ratcliffe 2008). Induction of the HIF-1α at low oxygen increases the transcription of glycolytic enzymes and induces ATP generation from glycolysis. HIF-1α induces the expression of pyruvate dehydrogenase kinase inhibiting pyruvate dehydrogenase activity, thereby blocking pyruvate decarboxylation to Acetyl-CoA and entry into the tricarboxylic acid cycle. This decreases mitochondrial respiration and ATP production by oxidative phosphorylation. Collective evidence suggests that HIF expression is an important adaption for neural survival under hypoxia (Taylor 2008).
9.2.6 Genes Associated with Cell Cycle Arrests
It is becoming increasingly evident that oxidative and nitrosative stresses are closely related with cell cycle abnormalities in neurons from patients with neurodegenerative diseases, supporting the view that these processes are interconnected and intertwined at the molecular level (Clopton and Saltman 1995; Mancuso et al. 2007; Kovacic and Somanathan 2012). Earlier studies on the effect of oxidative stress in the cell cycle reveal that increases in ROS-mediated DNA damage are correlated with cell cycle arrest (Clopton and Saltman 1995; Migliore and Coppede 2002). However, whether ROS-exposed cells undergo growth arrest or apoptosis may depend in part on where the cell resides in the cell cycle when insulted. For example, neural cells of neuronal and glial origin and human fibroblast treated with H2O2 undergo either cell cycle arrest or apoptosis. The majority of the apoptotic neurons and fibroblasts were in the S phase of the cell cycle, whereas growth-arrested fibroblast cultures were predominantly in the G1 or the G2/M phase (ElShamy et al. 1998; Chen et al. 2000). The mechanism by which oxidative and nitrosative stress may lead to cell cycle abnormalities remains unknown. However, it is proposed that increase in this ROS-mediated modification of DNA bases and strand breaks may be involved in cell cycle abnormalities associated with the pathogenesis of neurodegenerative diseases (Sekiguchi and Tsuzuki 2002).
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


