Fig. 10.1
Effect of diet and prolonged exercise-mediated oxidative stress, aging, and environmental and genetic factors on the onset of neurotraumatic and neurodegenerative diseases Plasma membrane (PM), glutamate (Glu), N-methyl D-aspartate receptor (NMDA-R), cytosolic phospholipase A2 (cPLA2), arachidonic acid (ARA), phosphatidylcholine (PtdCho), lysophosphatidylcholine (lyso-PtdCho), reactive oxygen species (ROS), cyclooxygenase-2 (COX-2), and platelet activating factor (PAF).
Neuropsychiatric disorders include both neurodevelopmental disorders and behavioral or psychological difficulties associated with some neurological disorders. Examples of neuropsychiatric disorders are schizophrenia, bipolar affective disorders, autism, mood disorders, manic depression, attention deficit disorder, dementia, tardive dyskinesia, and organic mental disorder. These disorders are closely associated with the abnormalities in cerebral cortex and limbic system (thalamus, hypothalamus, hippocampus, and amygdale) (Farooqui 2009, 2010).
The most important risk factors for stroke and neurodegenerative diseases are old age, positive family history, diet, sedentary lifestyle (lack of exercise), and endogenous and environmental factors (Fig. 10.1). It is becoming increasingly evident that mitochondria play an important role in the induction of oxidative stress and cumulative oxidative stress resulting in mitochondrial mutations, mitochondrial dysfunction, and oxidative damage (Lin and Beal 2006). However, as the association and role of ROS is increasingly recognized in aging and age-related diseases, number of questions are beginning to emerge in aging research. Is oxidative stress a secondary phenomenon of dysfunctional and dying neurons, or does oxidative stress itself cause the dysfunctionality/death of neurons? How does a global event such as oxidative stress result in the selective neuronal vulnerability observed in most neurodegenerative diseases? Finally, if oxidative stress is the primary cause for the pathogenesis of neurodegenerative diseases, then why treatment studies on the use of antioxidant therapy have largely failed?
As stated in Chap. 1, consumption of Western diet , which is enriched in n-6 fatty acids, high in cholesterol, sodium, refined carbohydrates, and low in fiber, increases the risk of developing neurotraumatic, neurodegenerative, and neuropsychiatric diseases (Farooqui 2009, 2010). In contrast, other types of diets, such as Mediterranean diet, traditional Japanese diet, ketogenic diet, and calorie restriction decrease the risk of developing above mentioned neurological disorders (Fig. 10.2). The onset of stroke and neurodegenerative diseases is often subtle and usually occurs in mid to late life and their progression depends not only on genetic, but also on environmental factors (Graeber and Moran 2002). The onset of neurological diseases may occur when neurons fail to respond adaptively to age-related increases in oxidative and nitrosative stress and neuroinflammation . Persistence presence of oxidative and nitrosative stress and neuroinflammation not only induces the accumulation of damaged proteins, DNA, and membrane fragments, but also alters transcriptional machinery both in aging and neurological disorders. In addition, neurodegenerative diseases are accompanied by the accumulation of disease-specific proteins, such as accumulation of Aβ and its aggregates in the cerebral cortex and hippocampal region in AD, α-synuclein in the brain stem in PD, and huntingtin in striatal medium spiny neurons in HD. Furthermore, abnormalities in signal transduction processes along with elevated levels of lipid mediators, and disturbance in stress resistance mechanisms have also been reported in neurological disorders (Farooqui 2010, 2011). Recent MRI studies have also indicated that despite differences in chemical composition and structures of the implicated proteins (Aβ, α-synuclein, huntingtin), the resulting dementias fall into dissociated but dispersed brain networks, supporting the view that the neurodegenerative diseases are transmitted along neuronal pathways rather than by proximity (Raj et al. 2012). Thus, atrophy in AD seems to spread in the brain along extant fiber pathways via secondary Wallerian degeneration, disconnection, loss of signaling, axonal reaction and post-synaptic dendrite retraction (Seeley et al. 2009). Based on network models of dementia progression, it is shown that atrophy in AD spreads from the hippocampus to the parietal regions connected to it, and eventually to the frontal connections, until the entire brain has been affected (Thompson et al. 2003). Accumulating evidence indicates that disease progression follows vulnerable fiber pathways rather than by proximity (Seeley et al. 2009; Englund et al. 1998; Villain et al. 2008). This understanding has led to a new conception of neurodegeneration as network diseases, affecting different parts of the brain differently (Buckner et al. 2005). Degeneration of neurons, loss of synapses, and abnormalities in fiber pathways in neurological disorders may cause problems with thinking, speaking, swallowing, breathing, skilled movements, decision making, cognition, and memory (Graeber and Moran 2002; Wishart et al. 2006; Soto and Estrada 2008; Farooqui 2010). Both stroke and neurodegenerative diseases lead to progressive cognitive and motor disabilities with devastating consequences to their patients. In older individuals and animals age-related alterations in interplay (cross-talk) among excitotoxicity , oxidative stress, and neuroinflammation may cause abnormalities in motor and cognitive performance. An enhanced rate (upregulation) of interplay among excitotoxicity, oxidative stress, and neuroinflammation may be a common mechanism of brain damage in stroke and neurodegenerative diseases (Farooqui and Horrocks 2007; Farooqui 2010). In addition, life style (diet, lack of exercise and sleep), genetic, and environmental factors (exposure to toxins in early life) may also be associated with the increased vulnerability of neurons in stroke, neurodegenerative, and neuropsychiatric diseases (Kidd 2005; Farooqui 2010).
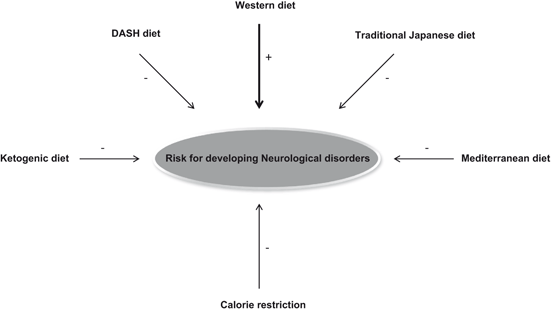
Fig. 10.2
Diet and risk for developing neurological disorders. Positive sign (+) indicates increased risk for developing neurological disorders where as negative sign indicates decrease in developing neurological disorders. It should be noted other factors, such as age, exercise, genetic, and environmental factors may also contribute to the onset of neurological disorders
10.2 Involvement of Oxidative Stress in the Pathogenesis of Neurological Disorders
Brain has an extraordinary high metabolic rate consuming approximately 20 % of all inhaled oxygen at rest; however, it only accounts for 2 % of body weight (Silver and Erecinski 1998) . This enormous metabolic demand is due to the fact that neurons are highly differentiated cells and need large amounts of ATP in order to maintain ionic gradients across cell membranes and for neurotransmission. Since most neuronal ATP is generated by oxidative metabolism, neurons depend critically on mitochondrial function and on oxygen supply (Kann and Liang 2007).
The mitochondria have critical functions which influence neuronal excitability, including the production of adenosine triphosphate (ATP), fatty acid oxidation, excitotoxicity , apoptosis and necrosis control, amino acid cycle regulation, biosynthesis of neurotransmitters, and regulating the homeostasis of cytosolic calcium. Mitochondria are the main site of ROS production therefore they are extremely vulnerable to oxidative damage (Waldbaum et al. 2010) .
It is known that a common feature of neurotraumatic, neurodegenerative, and neuropsychiatric diseases is the presence of oxidative and nitrosative stress. Oxidative and nitrosative stress (overproduction of superoxide anions, hydroxyl radicals, hydrogen peroxide, and peroxynitrite), which may be responsible for neuronal cell dysfunction and death in above mentioned neurological disorders (Barham et al. 2004; Maes et al. 2011; Farooqui 2010). At the molecular level, these reactive species are not only detrimental to neural cells through their oxidative effects, but also through the regulation of cellular redox, which plays an important role in the modulation of critical cellular functions (mainly in astrocytes and microglia), such as mitogene-activated protein (MAP) kinase cascade activation, ion transport, calcium mobilization, and apoptosis program activation (Emerit et al. 2004; Farooqui and Horrocks 2007; Farooqui 2010). Oxidative and nitrosative stress generating pathways are interconnected and closely associated with neuroinflammatory pathways and responses, along with subsequent involvement of mitochondrial metabolic processes, which generate more highly reactive free radical species. Indeed, ROS and reactive nitrogen species (RNS) consist of active moieties that can react with neural cell lipids, proteins, and nucleic acids. Under physiological conditions defense pathways counterbalance ROS and RNS production, thus in these conditions reactive species have physiological roles that include signaling neural cell proliferation, differentiation, and growth. Under pathological conditions excessive production of ROS or RNS may result in reactions of reactive species with fatty acids, proteins, and DNA, thereby causing damage to these cellular components (Barham et al. 2004; Maes et al. 2011) . Thus, the overproduction of ROS and RNS can damage all components of the cell, leading to a progressive decline in physiological function. As stated in Chaps. 7, 8, and 9, ROS can attack proteins causing their carbonylation, which is an irreversible oxidative damage, often leading to a loss of protein function and protein aggregation (Dalle-Donne et al. 2006). Generation of peroxynitrite results in the nitration of tyrosine residues in proteins (protein nitration), leading to alterations in protein activity (Ischiropoulos 2009). Free radicals can “steal” electrons from the lipids, often affecting polyunsaturated fatty acids, in the neural cell membranes (lipid peroxidation), resulting in degradation and peroxidation of lipids and cell damage. In addition, some end-products of lipid peroxidation, such as malondialdehyde, are mutagenic and carcinogenic (Nair et al. 2007). Furthermore, ROS can damage DNA, most readily at guanine residues, which causes mutations resulting in inheritable disease, cancer and aging (Cadet et al. 2003). In the past decades, these processes of oxidative damage have been well characterized in a large variety of diseases including neurological disorders .
10.2.1 Oxidative Stress in Stroke-Mediated Brain Injury
As stated above, stroke-mediated neuronal injury is accompanied by the interruption in oxygen supply, depletion in ATP generation, and mitochondrial dysfunction . The initial response to ATP depletion in ischemic injury is depolarization, which causes Na + influx into axons. Prolonged depletion of ATP produces a massive Ca 2 + influx and activation of phospholipases A2, proteases, and nitric oxide synthases that facilitate neurodegeneration (Farooqui and Horrocks 2007).
At the injury site, all vascular cells (endothelial cells, vascular smooth muscle cells, adventitial fibroblasts, and neurons) produce ROS primarily via cell membrane-bound NADPH oxidase (Sun et al. 2007). Other sources of ROS include uncontrolled arachidonic acid cascade and mitochondria, which generate significant levels of ROS during respiration. Oxidative stress occurs either from an excessive generation or decrease in clearance of ROS. Stroke also triggers a robust inflammatory reaction characterized by peripheral leukocyte influx into the cerebral parenchyma and activation of endogenous microglia (Iadecola and Alexander 2001; Allan et al. 2005; Farooqui and Horrocks 2007). It should be noted that even ischemic neurons, secrete inflammatory cytokines that cause, among other things, adhesion molecule upregulation in the cerebral vasculature which leads to peripheral leukocyte recruitment. Thus, neural cells are also capable of secreting chemokines, leading to further inflammatory cell chemotaxis into the ischemic lesion. Once activated, inflammatory cells can release a variety of cytotoxic agents including cytokines, matrix metalloproteinases (MMPs), nitric oxide (NO) and more ROS. In addition, oxidation of biogenic amines by monoamine oxidases generates hydrogen peroxide (H2O2), which in the presence of copper generates hydroxyl radicals (•OH). Neurons are particularly vulnerable to oxidative damage not only because of alterations in mitochondrial membrane potential (Atlante et al. 2000) but also due to inactivation of glutamine synthetase. Collective evidence suggests that multiple mechanisms contribute to neuronal injury and neural cell death following stroke-mediated brain injury (Farooqui 2010) .
It should be noted that stroke-mediated neuronal injury increases the prevalence of dementia by 10 %. An additional stroke increases the risk of dementia by another 10 % (Pendlebury and Rothwell 2009). Thus, the occurrence of a second stroke is a powerful predictor of dementia. The prevalence of cognitive impairment is even higher: 50 % of individuals exhibit deficits when assessed 3 months post-stroke (Sundar and Adwani 2010). Collective evidence suggests that stroke-mediated neuronal injury can therefore initiate rapid decline in cognitive function. This concept forms the basis of the pathogenesis of vascular cognitive impairment in dementia following stroke-mediated neuronal injury (Hachinski et al. 2006) .
10.2.2 Oxidative Stress in Traumatic Brain Injury
Multiple mechanisms contribute to secondary brain damage following TBI (Stoica and Faden 2010) . Oxidative stress, secondary to excitotoxicity and intracellular calcium dysregulation, plays a central role in secondary brain injury-mediated neuronal death (Petronilho et al. 2010; Vosler et al. 2009). Neurophysiological changes following TBI involve the initiation of an acute inflammatory responses, including breakdown of BBB, brain edema, infiltration of peripheral blood cells, mitochondrial dysfunction, and activation of resident immunocompetent cells, as well as the release of numerous immune mediators such as interleukins and chemotactic factors (Stahel et al. 2000). Oxidative stress begins with generation of ROS in the lesion area after TBI, leading to the initial production of superoxide (O2 ● −) and nitric oxide (●NO) radicals. ROS then react and metabolize to form stronger oxidants in the form of peroxynitrite (ONOO−), hydroxyl (● OH), carbonate (CO3 ● −), and nitrogen dioxide (● NO2) radicals, which in turn react with proteins, lipids, sugars, and nucleotides and impair the normal physiological function of cells (Lewen et al. 2000; Hall et al. 2004) . The generation of above reactive species initiates a cascade of neurochemical processes resulting in neuronal dysfunction and death, and thus playing a prominent role in the morbidity and mortality (Merenda et al. 2008). Mitochondrial dysfunction is consistently observed in brain tissue samples from patients with severe TBI (Verweij et al. 2000) and may serve as a key source for ROS and RNS production (Kagan et al. 2004, 2009). In an experimental model of pediatric TBI, selective oxidation of the mitochondrial lipid cardiolipin is observed very early after injury, supporting the view that mitochondria are an initial source of ROS and RNS production (Bayir et al. 2007). It should be noted that cardiolipin oxidation is intimately linked with the release of cytochrome c, oxidative stress may be critically linked to apoptotic neuronal death after TBI (Kagan et al. 2004, 2009). Importantly, enhancement of cellular neuroprotective mechanisms through the use of exogenous antioxidants has been shown to mediate neuroprotective effects in animal models of TBI (Wang et al. 2011; Hall et al. 2010) .
10.2.3 Oxidative Stress in Spinal Cord Injury
SCI is caused by an impact to the spinal cord . It consist of acute structural and physiological disruption of axons, neural cell damage and blood vessel ruptures. Bleeding and necrosis in the central gray matter at the lesion site occur within the first hours after the SCI (acute phase), followed by edema and more bleeding in the 6–7 h following trauma (Farooqui 2010). The injury is the result of ischemia that is caused by reduced blood flow to the affected spinal segment. This reduction in blood flow may not only be caused by a change in the spinal canal, but also by development in edema and bleeding along with reduction in systemic blood pressure. Ischemic injury creates a chain of neurochemical reactions (overstimulation of glutamate receptors, induction of a massive Ca2 + influx and activation of phospholipases A2, proteases, and nitric oxide synthases) resulting into neuronal cell death. Inflammatory cells then simultaneously migrate to the injured site with glial cell proliferation. The chronic phase lasts one to four weeks; during this time, the proliferation and hypertrophy of astrocytes form a glial scar or a cyst (Kakulas 1984; Farooqui 2010). Like stroke and TBI, pathogenesis of secondary injury in SCI involves abnormalities in several signal transduction mechanisms including enhancement in ROS and RNS producing pathways, which are activated not only through rise in intracellular calcium, but also by overstimulation of glutamate receptors, and release of cytokine and chemokines after the initial injury. The quick rise in ROS and RNS in cooperation with glutamate and cytokines enhances the oxidative stress propagation and onset of neuronal cell death. Early neuronal death is seen after SCI involves necrosis, while delayed neuronal death occurs via apoptosis (Springer et al. 1997). Now, there is substantial evidence that oxidative damage is a critical component of spinal cord trauma in rodent model of SCI (Aksenova et al. 2002; Huang et al. 2007; Farooqui 2010) .
10.2.4 Oxidative Stress in Alzheimer Disease
It is well known that oxygen is indispensable for life . Utilization of oxygen by mitochondria produces energy (ATP) as well as ROS and RNS. The generation of ROS and RNS results in alteration in cellular redox status. These species play a dual role as both toxic and beneficial compounds. Low or moderate levels, ROS and RNS exert beneficial effects on cellular responses and immune function, but at high concentrations, ROS and RNS generate oxidative stress, a deleterious process that can damage all cell structures (Valko et al. 2007; Pacher et al. 2007; Farooqui 2010). Oxidative damage has been implicated as an important mediator in the onset, progression and pathogenesis of AD. Redox-active metals, such as iron, are important causes of oxidative damage in AD because they accumulate in the brain of AD patients and are sources of redox-generated hydroxyl radicals(Sayre et al. 2000). In the brain of AD patients, excessive amounts of iron have been found to accumulate within the senile plaques and neurofibrillary tangles in the affected areas (Casadesus et al. 2004). Furthermore, iron can promote the cleavage and synthesis of Aβ precursor protein in an oxidative stress-mediated pathway (Tamagno et al. 2005) . Moreover, Aβ can be oxidatively modified by metal-catalyzed hydroxyl radicals and become more water-insoluble and resistant to the protease. The net result of enhancement in ROS and RNS production in AD includes advanced glycation end products (AGEs), nitration of tyrosine residues in proteins, formation of lipid peroxidation-mediated adduction products (4-hydroxynonenal, and acrolein) as well as carbonyl-modified neurofilament protein and free carbonyls (Farooqui 2010, 2011). Importantly, this damage involves all vulnerable neurons in nucleus basalis and hippocampal area of the brain (Farooqui 2010). These processes are accompanied by significant declines in antioxidants and antioxidant enzymes (glutathione, glutathione peroxidase, glutathione-S-transferase, and superoxide dismutase) supporting the view that production of high levels of ROS and RNS in AD brain may contribute to neurodegeneration. In addition, in neurodegenerative diseases oxidative stress, perturbed energy metabolism, and alterations of disease-related proteins also result in Ca2 + -dependent synaptic dysfunction and impaired plasticity. These processes may also lead to the neurodegeneration (Zundorf and Reiser 2011) .
10.2.5 Oxidative Stress in Parkinson Disease
In PD patients, ROS, such as hydrogen peroxide, are produced by dopamine redox chemistry through the Fenton reaction . Dopamine is a good metal chelator and electron donor, which reacts with iron and manganese to induce oxidative stress. An increase of metal iron(III) and total iron concentration has been shown to occur in the post-mortem substantia nigra of PD patients (Sofic et al. 1988;Kienzi et al. 1995). Furthermore, mutation in α-synuclein can promote the accumulation and interaction of dopamine with iron leading to enhancement in ROS production (Lotharius and Brundin 2002). Manganese can also cause oxidative damage to DNA, induce dopaminergic neuronal loss through apoptotic pathways, and reduce antioxidants such as GSH, catalase and thiols (Shen and Dryhurst 1998) .
It is well known that brain is rich source of polyunsaturated free fatty acids (PUFAs), which are highly susceptible to oxidants. Following oxidant-mediated damage to PUFAs, plasma and intracellular protein/lipid bilayer membranes can be profoundly affected. In PD, levels of PUFAs in the substantia nigra are decreased, while levels of malondialdehyde, a marker of lipid oxidation, are increased (Farooqui 2010, 2011). In addition, it is becoming increasing evident that there is marked increase in 4-hydroxynonenal, a lipophilic product of the peroxidation of membrane-bound arachidonic acid (Farooqui 2010, 2011). These observations are supported by the observation that the incubation of alpha-synuclein with 4-hydroxynonenal results in its covalent modification in which each molecule of the protein can incorporate up to six molecules of 4-hydroxynonenal (Qin et al. 2007). It is proposed that this irreversible modification of alpha-synuclein inhibits its normal fibrillation, perhaps by allowing the modified protein to adopt the protofibril conformation and cause a marked neurotoxicity to cultured dopaminergic neurons (Qin et al. 2007). Levels of other markers of nitrosative damage to proteins, such as free and bound nitrotyrosine and nitration and nitrosylation of proteins and especially of alpha-synuclein and parkin in PD are also increased in animal models of PD (Pennathur et al. 1999; Giasson et al. 2000; Chung et al. 2004). Collective evidence suggests that oxidative stress contributes to the pathogenesis and neurodegeneration in PD .
10.2.6 Oxidative Stress in Amyotrophic Lateral Sclerosis
ALS is a fatal neurodegenerative disorder characterized by progressive degeneration of motor neurons in the spinal cord, motor cortex and brainstem (Boillee et al. 2006) . Although the exact cause of neurodegeneration in ALS is not known, multiple pathophysiological mechanisms that trigger the loss of motor neuron have been proposed. These mechanisms include oxidative stress, mitochondrial impairment, axonal dysfunction, reactive astrocytosis, mutant superoxide dismutase expression, peroxynitrite toxicity, cytoskeletal disorganization, glutamate cytotoxicity, transcription dysfunction, inflammation, and apoptotic cell death (Farooqui and Horrocks 2007; Farooqui 2010). It is also suggested that synergistic interactions among excitotoxicity , oxidative stress, protein misfolding, and neuroinflammation may play a major role in pathogenesis of ALS (Shaw and Ince 1997; Farooqui and Horrocks 2007). Majority of ALS cases are sporadic. However, approximately 5–10 % cases of ALS are caused by familial inheritance (familial ALS, FALS). In about 20 % of FALS patients, there is mutation in the gene encoding the antioxidant enzyme Cu2 + /Zn2 + SOD (SOD1). In transgenic mice over expression of some of FALS-linked mutant SOD1 proteins results in the development of a neurological disorder that resembles ALS (Julien and Kriz 2006). The exact mechanism of oxidative and nitrosative stress in the pathogenesis of ALS is not fully understood. However, it is proposed that mutations in SOD1 may promote oxidative stress by weakening catalytic copper binding and buffering (Carri et al. 1994). Increase in NOS activity and elevation in levels of NOS protein have been observed in the motor neurons of amyotrophic lateral sclerosis (ALS) patients supporting a role of RNS in the ALS pathology (Abe et al. 1997). Higher levels of RNS can react with other free radicals such as superoxide and undergo complex reactions to form the strong oxidant ONOO− which causes cellular damage (Barber and Shaw 2010; Martinez and Andriantsitohaina 2009). In addition, disruption in redox regulation has also been implicated in the pathogenesis of ALS . Interestingly, several pathogenic mechanisms link ALS to redox-sensitive proteins, such as SOD1, and proteins whose active-site contain cysteine residues, such as protein disulphide isomerase (PDI) and thioredoxin (Guttamnn and Powell 2012). These proteins contain a thiol group which is highly sensitive to changes in redox conditions (Cooper et al. 2002; Akhtar et al. 2012). Even slight modulations in redox state are capable of producing neurotoxic species such as NO2, NO2 •, and ONOO− (Lipton et al. 1993), suggesting that redox stress could be of importance in disease (Barber and Shaw 2010). In mutant SOD1 transgenic models of ALS, treatment with various antioxidants result in delaying of disease onset and in some cases slow the progression of ALS (Turner and Talbot 2008). These observations support the view that oxidative and nitrosative stress contribute to the pathogenesis of ALS .
10.3 Relationship Between Exercise and Oxidative Stress
As stated in Chap. 1, exercise is defined as any planned structured activity, which leads to increase in energy expenditure and heart rate . Exercise is classified by the type, intensity, and duration of activity. There are different types of exercises in relation to intensity: (a) aerobic exercise is performed through using equipments, such as treadmill and bicycle or jogging, (b) Yoga, ancient Indian exercise, which involves physical postures, rhythmic stretching movements and regulated breathing, and (c) Tai Chi, ancient Chinese tradition that involves a series of movements performed in a slow, focused manner along with deep breathing. The aerobic metabolism primarily generates energy from fat, and with the use of oxygen it produces energy, without much accumulation of lactic acid in the blood. Yoga and Tai Chi are particularly useful for promoting flexibility of core muscles, maintenance of balance and fall prevention through induction of antidepressant-like effects such as modulation of neurotransmitter level, elevation in BDNF levels, and boosting serotonin along with reduction in inflammation, oxidative stress, blood levels of lipids and growth factors (Yang 2007; Saeed et al. 2010; Bussing et al. 2012). All types of exercises result in increased blood flow, hormesis , and mitochondrial biogenesis (Fig. 10.3). Thus, regular moderate physical exercise has been shown to produce many beneficial effects on the human health including marked decrease in mortality along with a reduced risk of developing cardiovascular and cerebrovascular diseases, neurological disorders, cancer, and diabetes (Blair et al. 2001; Oguma et al. 2002; Farooqui 2013) . Exercise produces cardiovascular and cerebrovascular changes and muscular fitness by elevating energy consumption, improving insulin sensitivity, increasing blood flow, strengthening the immune system, reducing inflammation, promoting sleep, and controlling weight. The molecular mechanisms underlying above mentioned processes are not fully understood. However, it is becoming increasingly evident that exercise not only improves the dyslipidemic profile by raising high density lipoprotein-cholesterol and lowering triglycerides in the body (Lakka and Laaksonen 2007), but also increases expression of GLUT4 and other proteins involved in insulin signaling and glucose metabolism (Houmard et al. 1993).
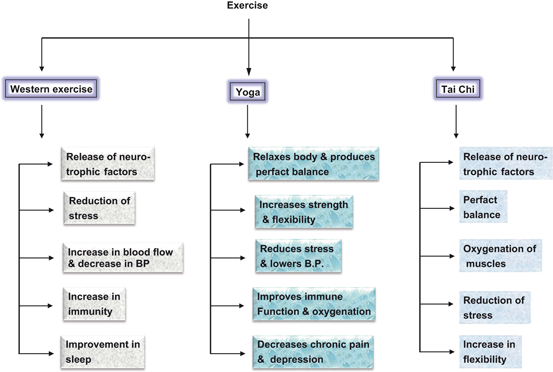
Fig. 10.3
Beneficial effects of aerobic exercise, Yoga, and Tai Chi on human health
Both resting and contracting skeletal muscles produce low levels of ROS and RNS through the involvement of mitochondria, NADPH oxidase, PLA2-dependent processes, xanthine oxidase, and increased expression of myokines (proteins secreted from skeletal muscle cells). Myokines include IL-6, IL-15, irisin, BDNF, MCP-1, ANGPTL-4, FGF-21, erythropoietin, and myonectin (Raschke and Eckel 2013). It is suggested that contraction-regulated myokines play a pivotal role in the communication between muscle and other tissues such as adipose tissue, liver, and pancreatic cells through the involvement of STAT, MAPK, PtdIns 3K/Akt and NF-κB signaling pathways (Pedersen 2011; Pedersen and Febraio 2008; Raschke and Eckel 2013). It is interesting to note that among above mentioned myokines, FNDC5, BDNFmRNA, erythropoietin-R mRNA, and their protein expression are increased in human skeletal muscle after exercise; however, muscle-derived BDNF and erythropoitin are not released into the circulation (Matthew et al. 2009). In muscle cells, BDNF not only increases phosphorylation of AMP kinase (AMPK) and Acetyl CoA Carboxylase (ACC), but enhances oxidation of fat both in vitro and ex vivo. In contrast, results on the involvement of erythropoietin have been inconsistent and inconclusive (Lamon and Russell 2013). AMPK is a key factor involved in modulation of exercise-mediated increase in fast-to-slow transition, a process that increases the proportion of oxidative fibers in the muscles (Narkar et al. 2008). AMPK is a heterotrimeric serine-threonine kinase, which consists of the catalytic α subunit (α1, α2), scaffolding β subunit (β1, β2) and nucleotide-binding γ subunit (γ1, γ2, γ3) (Steinberg and Kemp 2009). AMPK senses the AMP/ATP ratio via AMP binding to its γ subunit; AMP binding induces α subunit phosphorylation on Thr172 by AMPK kinases. Thr172 phosphorylation activates AMPK (Jensen et al. 2009). AMPK activation is associated with increased mitochondrial enzyme content and mitochondrial biogenesis in rat skeletal muscle. AMPK is activated by a low ATP/AMP ratio, and it has been proposed to serve as a fuel gauge for mammalian cells to protect against energy deprivation (Harding and Carling 1997). Acute activation of this enzyme in muscle helps defend against energy deficiency by promoting increased glucose transport and fatty acid oxidation through increased GLUT4 translocation (Bergeron et al. 2001) and inhibition of acetyl CoA carboxylase respectively (Atkinson et al. 2002). Mice lacking both AMPK β isoforms in skeletal muscles have drastically reduced exercise capacity, muscle mitochondria content and contraction-stimulated glucose uptake (O’Neil et al. 2011).
Recent studies have indicated that moderate exercise-mediated mild oxidative stress reversibly control the expression of at least 127 genes and signal transducing proteins reported to be not only sensitive to reductive and oxidative (redox) states in the cell (Allen and Tresini 2000; Frein et al. 2005), but also to gap junctional intercellular communication, which plays a role in coordinating gene expression between cells of a tissue needed to maintain cellular and tissue homeostasis (Upham and Trosko 2009). Moderate exercise-mediated changes in genes expression of skeletal muscles include alterations in expression of antioxidant enzymes, stress proteins, DNA repair proteins, and mitochondrial electron transport proteins (Allen et al. 2008; Powers and Jackson 2008). These proteins not only promote repair after injury, but also facilitate permanent beneficial adaptations, which may include increase in activators of mitochondrial biogenesis, such as the transcriptional coactivator peroxisome proliferator-activated receptor γ co-activator 1-α (PGC-1α) and increase in activity of oxidative enzymes such as citrate synthase and succinate dehydrogenase (Carter et al. 2001; Fernstrom and Fernstrom 2006). The molecular mechanisms that link PGC-1α with the control of ROS production remains unknown. However, it is known that PGC-1α activity is regulated by a variety of post-translational modifications including phosphsphorylation, acetylation, methylation and ubiquination (Jager et al. 2007; Rodgers et al. 2008; Dominy et al. 2010). Thus in neural (brain and spinal cord) and non-neural tissues (liver and muscles), PGC-1α is phosphorylated by p38 MAPK and AMPK at a variety of amino acid residues, which result in a more stable and active PGC-1α protein. The acetylase transferase (GCN5) acetylates PGC-1α at several lysine residues to inactivate PGC-1α while the NAD + -dependent deacetylase, sirtuin 1 (SIRT 1) removes acetyl groups leading to activation of PGC-1α (Fig. 10.4). SIRTs belong to Class III histone deacetylases, which regulate epigenetic gene silencing and suppress recombination of rDNA (Yamamoto et al. 2007). In mammals, SIRTs have a range of molecular functions and have emerged as important proteins in aging and metabolic regulations (Yamamoto et al. 2007; Rajendran et al. 2011). SIRTs represent a small gene family with seven members designated as SIRT1–7, known to be modulated by oxidative stress (Yamamoto et al. 2007; Rajendran et al. 2011) . Accumulating evidence suggests that post-translational modifications activate and/or deactivate PGC-1α located in the nucleus or within the cytosol of the cell. Recent studies have also indicated that moderate exercise activates a cytosolic pool of PGC-1α and promotes its migration to the nucleus, where it initiates mitochondrial gene expression prior to increase in overall PGC-1α expression. Thus, post-translational modifications represent an immediate mechanism to activate PGC-1α and initiate PGC-1α-dependent gene expression (Wright et al. 2007). PGC-1α has a powerful suppressive effect on ROS production, in parallel to its effects in elevating mitochondrial respiration. This occurs through the PGC-1α-mediated expression of genes involved in ROS detoxification, as well as the expression of uncoupling proteins that can attenuate ROS production (St-Pierre et al. 2006; Valle et al. 2005). In addition, heat shock proteins (Hsps), which are induced under oxidative stress, promote the production of anti-inflammatory cytokines, indicating immunoregulatory potential of these proteins. Therefore, the presence of immune responses to Hsps in inflammatory diseases is considered as an attempt of the immune system to correct the inflammatory condition (Lakka and Laaksonen 2007; Morton et al. 2009). It is also proposed that exercise boosts the production of human growth hormone and blocks maladaptive pathways, which may not only provide protection against protein misfolding diseases, but also promote preservation of muscle function during aging (Morton et al. 2009). Recent studies have also indicated that human genome contains genes controlling either the motivation or ability to exercise (Good et al. 2008). Collective evidence suggests that exercise-mediated production of low levels of ROS plays an important physiological function in the regulation of both muscle force production and contraction-induced adaptive responses of muscle fibers . These processes not only involve the participation of myokine-mediated signal transduction pathways, but also mediate changes (a) transcriptional regulation of nuclear-encoded genes encoding mitochondrial proteins by the PGC-1, (b) control of mitochondrial DNA gene expression by the transcription factor Tfam, (c) mitochondrial fission and fusion mechanisms, and (d) import of nuclear-derived gene products into the mitochondrion via the protein import machinery. Exercise can modify the rates of various steps involved in the biogenesis of mitochondria. Muscle mitochondrial biogenesis involves the assembly of an interconnected network system (mitochondrial reticulum). This expansion of membrane size is modulated by the balance between mitochondrial fusion and fission. Accumulating evidence suggests that mitochondrial biogenesis is an adaptive mechanism, which requires the coordination of multiple cellular events, including the transcription of two genomes, the synthesis of lipids and proteins and the stoichiometric assembly of multisubunit protein complexes into a functional respiratory chain. Impairment at any step may not only cause defective electron transport, but also result in a subsequent failure of ATP synthesis, leading to an inability to maintain redox potential and energy homeostasis (Hood et al. 2006) .
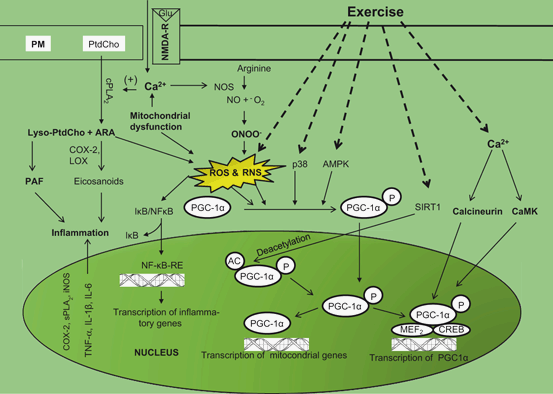
Fig. 10.4
Diagram showing activities and activation of NF-κB and PGC-1α following exercise in the brain. Plasma membrane (PM), glutamate (Glu), N-methyl D-aspartate receptor (NMDA-R), cytosolic phospholipase A2 (cPLA2), arachidonic acid (ARA), phosphatidylcholine (PtdCho), lysophosphatidylcholine (lyso-PtdCho), reactive oxygen species (ROS), cyclooxygenase-2 (COX-2), lipoxygenase (LOX), platelet activating factor (PAF), reactive nitrogen species (RNS), peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1), nuclear factor-κB (NF-κB), nuclear factor-βB-response element (NF-κB-RE), inhibitory subunit of NF-κB (I-κB); tumor necrosis factor-α (TNF-α); interleukin-1β(IL-1β); interleukin-6 (IL-6), inducible nitric oxide synthase (iNOS), secretory phospholipase A2 (sPLA2), nitric oxide (NO), peroxynitrite (ONOO – ); p38 MAP kinase (p38), AMP-activated protein kinase (AMPK), cAMP response element binding protein (CREB), EBox binding proteins (MEF2), and NAD+-dependent protein deacetylase (SIRT1), Ca2+/calmodulin-dependent protein kinase (CaMK)
In contrast, intense and prolonged exercise results in high ROS and RNS-mediated oxidative damage to both proteins, release of excess of cortisol , and lipid degradation in the contracting myocytes. It is suggested that high levels of ROS and RNS promote contractile dysfunction resulting in muscle weakness and fatigue (Fig. 10.5). The molecular mechanisms associated with muscle contractile dysfunction are not fully understood. However, it is becoming increasingly evident that high levels of oxidants (ROS and RNS) along with high levels of cortisol may alter cell signaling pathways, which are closely associated with contractile dysfunction. Thus, ROS and RNS have been reported to modulate a number of cell signaling pathways as well as regulate the expression of multiple genes in eukaryotic cells (Allen et al. 2008; Powers and Jackson 2008). ROS and RNS-mediated changes in gene expression not only involve changes at transcriptional level and stability of mRNA, but also intensity of signal transduction processes involved in muscle contraction leading to fatigue and microscopic tears in the skeletal muscles, increase in inflammation in lining of arteries, and sudden cardiac fatigue promoted by increase in catecholamines and adrenalin, which may trigger arrhythmia and cardiac arrest (Fig. 10.6) . One common arrhythmia is atrial fibrillation, commonly known as “A-fib”. A-fib is epidemic among endurance athletes, which sets them up for major increase in stroke risk. Marathoners above age 50 have a five-fold increase in A-fib rates (O’Keefe et al. 2012) supporting the view that high intensity running is harmful for human health. High levels of cortisol may not only promote weight gain (visceral obesity) and sleeping problems, but may also induce obesity (weight gain). Some studies have shown that elevated cortisol levels tend to cause fat deposition in the abdominal area rather than in the hips (Tchernof and Despres 2013). This fat deposition has been referred to as “toxic fat” since abdominal fat deposition is strongly correlated with the development of cardiovascular disease including heart disease and strokes (Farooqui 2013) .
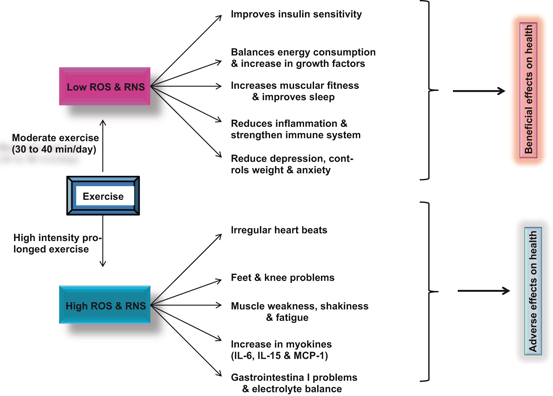
Fig. 10.5
Effect of moderate and intensive prolonged exercise on ROS production. Reactive oxygen species (ROS), reactive nitrogen species (RNS), interleukin-6 (IL-6), inteleukin-15 (IL-15), and monocyte chemoattractant protein-1 (MCP-1)
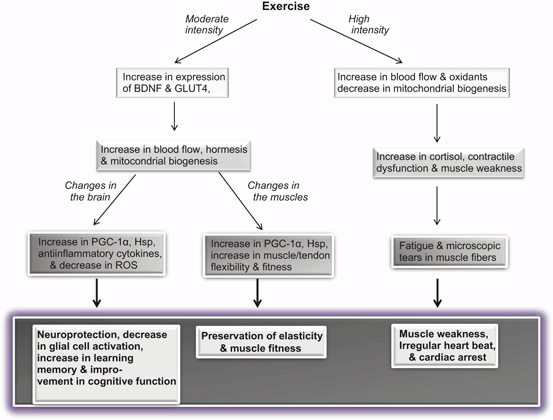
Fig. 10.6
Effect of moderate and high intensity exercise on muscles and brain. Brain-derived neurotrophic factor (BDNF); Peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC–1); heat shock protein (Hsp); Glucose transporter type 4 (GLUT4); and reactive oxygen species (ROS)
10.4 Effect of Exercise, Hormesis, and Other Neurochemical Parameters in the Brain
Moderate physical exercise is known to produce preventive and therapeutic effects in neurological disorders (Deslandes et al. 2009; Wichi et al. 2009) . The neuroprotective effect of exercise can be explained by the hormesis, which is defined as a biphasic dose response whereby moderate exercise stimulates resistance to stress and improves biological fitness, while too much exercise induces damage and inhibits function. This concept of hormesis can be extended to the effect of exercise on ROS and RNS production in muscles (Radak et al. 2005; Radak et al. 2008). ROS and RNS generated during exercise can cause oxidative and nitrosative stress and damage to skeletal muscle, heart muscle, and muscles of other organs, and neurochemical changes in the brain. It is well known that at low levels ROS and RNS serve as the chemical agents (mediators) for maintaining cellular milieu and transferring messages from one subcellular organelle to another to conduct physiological functions, such as contraction, bioenergetics, growth, proliferation and remodeling (adaptation). The preventive and therapeutic effects of regular exercise, at least in part, can be explained by ROS and RNS-mediated adaptation. The oxidative challenge-related adaptive processes regulated by exercise do not just depend upon the production of ROS and RNS but also on the increase in antioxidant and housekeeping enzyme activities, which are associated with modulation of the oxidative damage repairing enzymes. Thus, moderate exercise-induced ROS and RNS production plays an important role in the induction of antioxidants, induction of Hsps, DNA repair, and protein-degrading enzymes and significantly reduce incidence of oxidative stress induced by the disease. Exercise also mediates it effects by increasing the circulation of same proinflammatory cytokines (myokinase) that are normally upregulated during a mild stress response. However, as stated above, exercise may also upregulate the expression of anti-inflammatory cytokines, which inhibit inflammation and with time increase the immune system threshold for stress (Zaldivar et al. 2006). Studies on RNAs from rodents exposed to a running wheel for 3, 7 and 28 days using a microarray indicate that exercise mediates the expression of 1176 cDNAs in the brain (Molteni et al. 2002) . Quantification of selected genes by Taqman RT-PCR or RNase protection assay indicates the onset of up-regulation of genes associated with synaptic trafficking (synapsin I, synaptotagmin and syntaxin); signal transduction pathways (Ca2 + /calmodulin-dependent protein kinase II, CaM-KII; mitogen-activated/extracellular signal-regulated protein kinase, MAP-K/ERK I and II; protein kinase C, PKC-delta) or transcription regulators (cyclic AMP response element binding protein, CREB) (Molteni et al. 2002). In addition, genes associated with the glutamatergic neurotransmission (N-methyl-d-aspartate receptor, NMDAR-2A and NMDAR-2B and excitatory amino acid carrier 1, EAAC1) are also up-regulated, while genes related to the gamma-aminobutyric acid (GABA) neurotransmission are down-regulated (GABAA receptor, glutamate decarboxylase GAD65). The temporal profile of exercise-mediated gene expression delineates a mechanism by which specific molecular pathways are activated after exercise performance. For example, the CaM-K signal system is turned on and activated during acute and chronic periods of exercise, while the MAP-K/ERK system play an important role moderate during long-term exercise sessions (Molteni et al. 2002). At the molecular level moderate exercise initiates and stimulates intracellular programs that not only upregulate antioxidant systems (copper–zinc (CuZn) and manganese (Mn) superoxide dismutase (SOD1 and SOD2, respectively), which facilitate repair, but also promotes mitochondrial biogenesis. Several transcription factors, enzymes, and chaprone proteins such as nuclear factor erythroid 2-related factor 1/2 (Nrf-1/2), PPAR γ coactivator 1 (PGC-1), forkhead transcription factors (FOXO), sirtuins, AMPK, mitochondrial transcription factor A (Tfam) and Hsps contribute to the beneficial effects of exercise (Figs. 10.7 and 10.8) (Calabresse et al. 2010; Sano and Fukuda 2008; Son et al. 2008). Among these factors, Hsps play an important role in neuroprotection. Hsps are induced via highly regulated signaling cascades, including the three major mitogen-activated protein kinases (MAPK) and protein kinase B (PKB/Akt) (Nadeau and Landry 2007; van Ginneken et al. 2006; Wigmore et al. 2007) . Both these pathways are impacted by exercise (Chen and Russo-Neustadt 2005; Shen et al. 2001). The exercise-mediated alterations in MAPK element p38 not only upregulate phosphorylation of small heat shock protein (sHsp, mol mass 15–42 kDa) (Ito et al. 2005; Maizels et al. 1998), but also down-regulate pAkt and pERK activities, which may promote dephosphorylation in HSF1-ser307 leading to stabilization of its activity (Seo et al. 2006; Wigmore et al. 2007). Based on this information, it is suggested that voluntary moderate exercise upregulates the small heat shock proteins Hsp27 and produces elevation in the pre-synaptic and SNAP-25 and the post-synaptic protein PSD95, which coincides with the sHsp response (Fig. 10.9). The transport of PSD95 to dendrites through PtdIns 3K-induced Akt signaling is coupled with BDNF-mediated activation of NMDA receptor and influx of Ca2 + (Yoshii and Constantine-Paton 2007). In addition, exercise also induces immediate early gene (IEG) along with IEGs, which are induced preferentially by depolarization (IPD-IEG) (Machado et al. 2008). These processes not only play an important role in neuroprotection via the production and secretion of BDNF and Hsps (Hu et al. 2009; Vaynman et al. 2004; Vaynman et al. 2006). Exercise also decreases blood pressure by attenuating oxidative stress in the paraventricular nucleus (PVN) and rostral ventrolateral medulla (RVLM) of Spontaneously hypertensive rats (SHRs), possibly by reducing sympathoexcitation supporting the view that chronic exercise not only attenuates proinflammatory cytokines and the vasoconstrictor axis of the brain/central renin-angiotensin system (RAS) but also attenuates sympathoexcitation, improves anti-inflammatory defense mechanisms and vasoprotective axis of the RAS in the brain, which, at least in part, explains the blood pressure-lowering effects of exercise in hypertension (Agarwal et al. 2011) .
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


