Fig. 4.1
Cytokines and chemokines associated with neuroinflammation
In addition, stimulation of glutamate, cytokine, Toll-like, bradykinin, histamine, dopamine, purinergic, GABA, endothelin, and platelet activating factor (PAF) receptors results in stimulation of phospholipid and sphingolipid degrading enzymes including phospholipases A2(PLA2) , cyclooxygenase-2 (COX-2) , inducible nitric oxide synthase (iNOS), superoxide dismutase (SOD), NADPH oxidase, and sphingomyelinases (SMases) (Kettenmann et al. 2011) (Fig. 4.2). Activation of these enzymes leads to the generation of high levels of eicosanoids , quinolinic acid, reactive oxygen species (ROS) , and nitric oxide (NO), which cause neuronal dysfunction and cell death (McGeer and McGeer 1995; Lai and Todd 2006; Farooqui 2010a). Microglial cells can recognize a wide range of signals that can threaten the structural and functional integrity of neuronal cell through various receptors (Kettenmann et al. 2011). Traumatized neurons release ATP and chemokine CXC motif ligand 10 (CXCL10), which attract microglia via activated purinoreceptors (Davalos et al. 2005; Haynes et al. 2006) and chemokine CXC motif receptor 3 (CXCR3), respectively (Rappert et al. 2004). In addition, microglial cells have also been reported to sense neuronal activity through neurotransmitter receptors that are present on the microglial cell plasma membrane (Pocock and Kittenmann 2007). For example, microglial cells contain glutamate receptors that link neuroinflammation and oxidative stress with excitotoxic brain damage (Pocock and Kittenmann 2007; Tahraoui et al. 2001) supporting the view that microglial cells are closely associated with the pathogenesis of neurodegeneration. The inhibition of microglial activation results in the attenuation of neurotoxic events leading to neuronal survival.
Fig. 4.2
NF-κB mediated stimulation of enzymes involved in neuroinflammation
Amongst the numerous factors released by activated microglial and astroglial cells, excessive NO● production induces neuronal cell death by damaging the mitochondrial electron transport chain function in neurons leading to disruption in neuronal ATP synthesis and increase in generation of ROS (Moncada and Bolanos 2006). Activation of NADPH oxidase in microglia results in generation of both superoxide (O2 ●−) and releases proinflammatory TNF-α (Qin et al. 2004). NO● produced in microglia or astrocytes may react with O2 ●− to generate the neurotoxic peroxynitrite radical (ONOO−) (Bal-Price et al. 2002). ONOO− inhibits mitochondrial respiration, induces caspase-dependent neuronal apoptosis, and promotes glutamate release resulting in excitotoxicity and neuronal death (Bal-Price et al. 2002; Abramov et al. 2005; Brown and Bal-Price 2003). Thus, the induction of neuroinflammation not only involves interplay among microglia, astrocytes, neurons, PMN, and endothelial cells but also interactions among various proinflammatory mediators that derived from enzymic and nonenzymic degradation of phospholipids, sphingolipid, and cholesterol (Farooqui et al. 2007). In addition, receptors (glutamate receptors, eicosanoid receptors, and toll-like receptors), transcription factors (nuclear factor-kappaB (NF-κB), bradykinin, and histamine receptors), increased expression of proinflammatory cytokines (TNF-α, IL-1β, and IFN-γ) and chemokines (MCP1and CXCL3) also contribute to the onset and maintenance of neuroinflammation. Thus, neuroinflammation has two sides. On one hand neuroinflammation may be beneficial in promoting homeostasis and neuronal survival, but on the other hand it can also result in tissue injury through the over-production of inflammatory mediators (PGs, LTs, TXs, PAF, TNF-α, IL-1β, and IFN-γ) .
It must be mentioned here that most of above mentioned results have been obtained on standard cultured microglial cells, which suffer from several disadvantages: (a) standard microglial cells are derived from the neonatal brain. Cultured microglial cells do not undergo maturation process that occurs in vivo, (b) cultured microglial cells are grown in serum-containing (usually 10 %) medium, whereas in vivo microglia normally never come in contact with serum components, and (c) under in vivo conditions, microglial cells are under constant restraint by a variety of inhibitory chemokines and immunomodulatory molecules such as CX3CL1, CD200, CD22, or CD172 (Biber et al. 2007; Prinz and Mildner 2011). This does not occur in cultured microglial cells. In addition, following infection or brain trauma peripheral monocytes/macrophages enter the brain. It is stated that the genetic removal of even just one chemokine or immunomodulatory molecule in animal models may dramatically change the reaction profile of microglia, often causing overshooting microglia reactions and sometimes even toxic microglia responses (Cardona et al. 2006); therefore, it is possible that the complete lack of normal inhibition may produce a dramatic influence on the reactivity of cultured microglial cells (Hellwig et al. 2013; Melief et al. 2012).
4.2 Contribution of Glutamate Receptors, Toll-like Receptors, and Eicosanoid Receptors in Neuroinflammation
Overstimulation of NMDA type of glutamate receptors is accompanied by calcium influx that initiates a cascade of events involving mitochondrial dysfunction, activation of enzymes associated with the release and oxidation of arachidonic acid (ARA) , and generation of reactive oxygen species (Farooqui and Horrocks 1994; Farooqui and Horrocks 2006). These enzymes include isoforms of PLA2 , cyclooxygenase-2 (COX-2) , lipoxygenases (LOX) , and epoxygenases (EPOX) (Phillis et al. 2006) (Fig. 4.3). An uncontrolled and sustained increase in cytosolic calcium levels also contributes to the stimulation. calpains, nitric oxide synthase, protein phosphatases, and various protein kinases (Arundine and Tymianski 2004; Farooqui et al. 2008). Accumulation of oxygenated ARA metabolites and elevation in platelet activating factor (PAF) along with abnormal ion homeostasis, changes in redox status, and lack of energy generation is associated with neural cell injury and cell death in acute neural trauma (ischemia, epilepsy, head injury, and spinal cord trauma) and neurodegenerative diseases such as Alzheimer disease (AD), and Parkinson disease (PD) (Farooqui et al. 2007; Farooqui et al. 2008) .
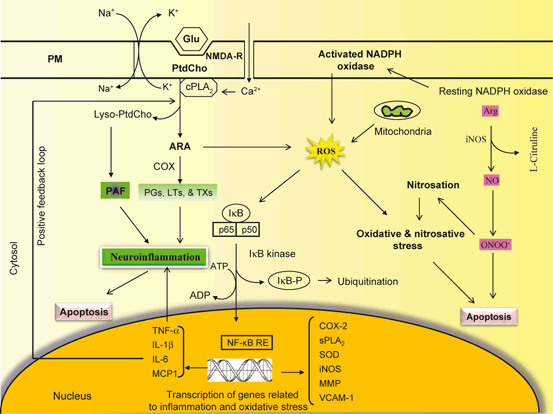
Fig. 4.3
Molecular mechanism associated with NF-κB-mediated transcription of genes involved in neuroinflammation. Glu Glutamate, NMDA-R NMDA receptor, PLA 2 phospholipase A2, PtdCho phosphatidylcholine, lyso-PtdCho lyso-phosphatidylcholine, ARA arachidonic acid, COX cyclooxygenase, PGs prostaglandins, LTs leukotrienes, TXs thromboxanes, PAF platelet activating factor, ROS reactive oxygen species, NF-kB NF-kappaB, NF-kB-RE NF-kappaB response element, I-κB inhibitory subunit of NF-κB, TNF-α tumor necrosis factor-alpha, IL-1β interleukin-1beta, IL-6 interleukine-6, MCP1 monocyte chemotactic protein-1, COX-2 cyclooxygenasse-2, iNOS inducible nitric oxide synthase, NO nitric oxide, ONOO − peroxynitrite, SOD superoxide dismutase, MMP matrix metalloproteinase, VCAM vascular adhesion molecule-1, sPLA 2 secretory phospholipase A2
Stimulation of NMDA receptors increases the generation of prostaglandins, leukotrienes, and thromboxanes from ARA. These metabolites are collectively called as eicosanoids (Phillis et al. 2006). Neurons and glia generate prostaglandins, whereas cerebral microvessels and the choroid plexus mainly produce thromboxanes. Eicosanoids act through their receptors and modulate signal transduction pathways and gene transcription. Thus, PGD2 activates the Prostaglandin D2 receptor (DP receptors), PGE2 activates the PGE2-activated receptors (EP receptors), and PGF2α, PGI2, and TXA2, respectively, stimulate the Prostaglandin F2α receptor, PGI2 Receptor and Thromboxane receptor (FP, IP, and TP receptors) (Coleman et al. 1994; Phillis et al. 2006). These receptors are linked to the generation of cyclic AMP, diacylglycerol, and phosphatidylinositol 1,4,5-trisphosphate and modulation of Ca2+ influx. Availability of glutamate in the synaptic cleft increases neuronal excitability and synaptic transmission at the presynaptic level whereas the uptake of glutamate by astrocytes prevents glutamate accumulation in the synaptic cleft and retards glutamate-mediated neurotoxicity. Thus, interplay among glutamate, prostaglandin, leukotriene, and thromboxane receptors in the brain may modulate neuronal cell survival. Under normal conditions, this cross talk refines the communication among receptors, but under pathological situations, the interplay among prostaglandin, leukotriene, and thromboxane receptors may promote neuronal injury, which depends upon the levels of glutamate, magnitude of PLA2 expression, levels of eicosanoids and the intensity of neuroinflammation and oxidative stress (Farooqui and Horrocks 2006; Farooqui et al. 2008). In addition, glutathione, a tripeptide involved in maintaining redox status binds to its own receptors and modulates glutamatergic excitatory neurotransmission by displacing glutamate from NMDA receptors (Janaky et al. 1999) resulting in reduction of reduced glutathione. Collective evidence suggests that in neural cells, glutamate-mediated modulation of eicosanoid generation, activities of glutamate transports, depletion of glutathione, and decreased ATP levels are closely associated with neuroinflammation and oxidative stress-mediated neuronal cell death (Gilroy et al. 2004; Manev et al. 2000) .
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are characterized by an extracellular leucine-rich repeat domain and an intracellular Toll/IL-1 receptor (TIR) domain. TLRs recognize a variety of highly conserved structural motifs expressed by microbial pathogens, called pathogen-associated molecular patterns (PAMPs) (Kaisho and Akira 2004). Mammals contain at least ten TLRs. Despite a high degree of structural similarity, each receptor has a distinct function in innate immune recognition. Each TLR has some degree of ligand specificity, which specificity is extended through dimerization of TLRs. Some TLRs employ additional co-receptors that assist in pathogen recognition, such as CD14 for TLR4 (Kielian 2006). In the brain tissue, Toll-like receptors (TLRs) are mainly expressed in microglial cells (Kettenmann et al. 2011) . TLRs are essential for mounting an immune response against infection. Thus, TLRs serve as pathogen-associated molecular pattern recognition receptors that bind microbial molecular motif with high affinity, and play a central role in the initiation of cellular innate immune responses (Aravalli et al. 2007; Kawai and Akira 2010). The stimulation of TLRs in microglial cells triggers the release of several cytokines and chemokines (Aravalli et al. 2007; Okun et al. 2009). Thus, detailed investigations have indicated that the stimulation of TLR3 causes the release of IL-6, IL-12, chemokine ligand 10, TNF-α , and IFNβ (Alexopoulou et al. 2001; Jack et al. 2005); TLR2 modulates the secretion of IL-6 and IL-10 (Jack et al. 2005); TLR9 regulates the production of NO and TNF-α (Iliev et al. 2004), and stimulation of TLR4 triggers the release of IL-6 and TNF-α (Okun et al. 2009). Most of above mentioned cytokines and their receptors (see below) are associated with the release of ARA and ceramide from neural membrane phospholipids and sphingolipid through the activation of PLA2s and SMases leading to the generation of proinflammatory lipid mediators (eicosanoids, platelet activating factor, and ceramide) (Farooqui 2011). As stated above, the cytoplasmic domains of the TLRs share a high degree of homology with the intracellular portion of the IL-1R (Takeda et al. 2003). TLR activation results in the recruitment of the adaptor protein MyD88, which is associated with the serine/threonine kinase IL-1R-associated kinase (IRAK). IRAK then interacts with the tumor necrosis factor (TNF) receptor-associated factor (TRAF) adaptor protein TRAF6, which provides a bridge to the protein kinase NF-κB-inducing kinase (NIK) (Fig. 4.4). NIK then phosphorylates IKK (IκB kinase), resulting in the phosphorylation of IκB. As stated above, IκB phosphorylation targets the protein for ubiquitination and proteasome-mediated degradation, resulting in the release and nuclear translocation of NF-κB, where it can influence the expression of numerous immune response genes (Kielian 2006) .
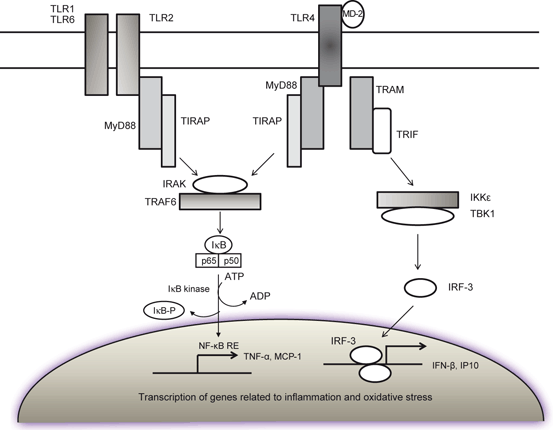
Fig. 4.4
Contribution of TLR receptors in cytokines and chemokine production during neuroinflammation. TLR2 and TLR4 Toll-like receptors 2 and 4, MyD88 adaptor protein, IRAK IL-1R-associated kinase, TRAF6 tumor necrosis factor receptor-associated factor adaptor protein 6, NIK NF-κB-inducing kinase, IKK IκB kinase, TRIF TIR-domain-containing adapter-inducing interferon-β, IFN-β interferon-beta, IRF-3 interferon regulatory transcription factor-3, NF-kB NF-kappaB, NF-kB-RE NF-kappaB response element, I-κB inhibitory subunit of NF-κB, TNF-α tumor necrosis factor-alpha, IP-10 IFN inducible protein of 10 kDa, MCP1 monocyte chemotactic protein-1
Eicosanoids are released outside of the cells immediately after their synthesis. They act through eicosanoids receptors that are located on plasma and nuclear membranes. Eicosanoid receptors include EP1, EP2, EP3, and EP4. These receptors are encoded by different genes and differ in their responses to various agonists and antagonists. Among eicosanoid receptors, EP2 activation is involved in microglial-mediated paracrine neurotoxicity as well as suppression of microglia internalization of aggregated neurotoxic peptides along with neuroinflammation (Coleman et al. 1994; Cimino et al. 2008). In addition, brain also contains PGF receptor (FP), PGI receptor (IP), and TXA receptor (TP), which may contribute to neurotransmitter release, sleep, and vasodialation and vasoconstriction of cerebral vessels in the brain .
RAGE is a type I transmembrane protein composed of three extracellular immunoglobulin-like domains (V, C1, and C2), a single transmembrane domain and a short cytoplasmic tail thought to be important in signal transduction (Hudson et al. 2008). RAGEs are expressed in the brain in neurons, microglia, and astrocytes (Lue et al. 2001; Sasaki et al. 2001; Yan et al. 1996). Aβ is a specific ligand for RAGE, which interacts with the N-terminal domain of RAGE (Chaney et al. 2005). RAGE expression is elevated in AD pathology-enriched brain regions, including hippocampus and inferior frontal cortex, when compared to cerebellum where AD pathology is limited. The interactions of RAGE with its diverse ligands (AGEs, Aβ, amphoterin and S100B protein) mediate multiple physiological and pathological functions, including inflammation, oxidative stress, neurodegeneration, maintenance of homeostasis, tumorigenesis, promotion of neurite outgrowth, cell survival, cell migration, and neuronal differentiation (Ramasamy et al. 2005; Meneghini et al. 2010). The binding of AGEs with RAGE not only results in the activation of the mitogen-activated protein kinases (MAPKs) and the phosphatidylinositol-3 kinase (PtdIns 3K) pathways, but also in activation of NADPH oxidases, generation of superoxide, and activation of NF-κB. This transcription factor migrates to the nucleus and induces the induction of inflammatory cytokines and adhesion molecule 1 (Ramasamy et al. 2005; Meneghini et al. 2010) .
4.3 Contribution of Transcription Factors in Neuroinflammation
NF-κB is a family of transcription factors that regulates multiple cellular functions associated with immune responses, neuroinflammation , apoptosis, cell growth and survival (Celec 2004). The mammalian NF-κB family members include NF-κB1 (p105 and p50), NF-κB2 (p100 and p52), RelA (p65), RelB and c-Rel, which is activated by upstream signals from diverse immune receptors, such as ligand-triggered Toll-like receptors (TLRs) , interleukin-1 receptor (IL-1R), tumor necrosis factor receptor (TNF-R) (see below) and antigen receptors (Hayden and Ghosh 2004; Rahman et al. 2009). The activation of NF-κB is tightly regulated by inhibitors of NF-κB (IκBs). These proteins are also involved in the regulation of these diverse cellular processes, such as neuroinflammation, apoptosis, cell growth and survival (Baeuerle and Baltimore 1988). In unstimulated cells, NF-κB proteins are localized in an inactive in the cytoplasm, usually as homodimers or as heterodimers with RelA, RelB or c-Rel and complexed with the inhibitory IκBs (Figs. 4.5 and 4.6). The IκBs are in turn regulated by another group of regulatory proteins called IκB kinases (IKKs). The IKK complex is composed of two catalysis subunits (IKKα and IKKβ) and a regulatory subunit IKKγ, or NEMO (NF-κB essential modulator) (Perkins 2007). Upon ligand engagement, the receptors initiate signal transduction events that lead to the activation of IKK complex by activated cellular protein kinases like NF-κB-inducing kinase (NIK), mitogen-activated protein/extracellular signal-regulated kinase1 (MEKK1), transforming growth factor-β (TGFβ)- activated kinase 1 (TAK1), MEKK2 or MEKK3. Activated IKK phosphorylates IκBα using specific serine residue within the IκBα proteins, triggering their ubiquitination via ubiquitin ligase (Traencker et al. 1995) . The IκBα protein is then degraded by the 26S proteasome, allowing the release and translocation of the active NF-κB dimer into the nucleus, where it binds to target sequences in the genome and facilitates the expression of a number of proteins including many enzymes (sPLA2, COX-2, NADPH oxidase and inducible nitric oxide synthase) and cytokines (TNF-α, IL-1β, and IL-6) (Figs. 4.1and 4.2) (O’Neill 2003; Karin and Ben-Neriah 2000). Collective evidence suggests that NF-κB can be activated by distinct mechanisms. These include microbial invasion recognized by TLR, generation of ROS, cellular generation of inflammatory eicosanoids , and interaction with inflammatory cytokines via defined cell surface receptors (see below). It is also known that several of these initiating events are modulated by dietary factors. This also means that appropriate use of the diet can either turn on or turn off the activation of NF-κB. This new knowledge is the foundation of anti-inflammatory nutrition (Sears 2008; Sears and Riccordi 2011) . Antioxidant, M40403, not only blocks excitotoxicity, but also prevents NF-κB translocation to the nucleus (McInnis et al. 2002). NMDA receptor-mediated activation of NF-κB involves ΙκB-α degradation by a caspase-3-like cysteine protease. In addition to NF-κB, other transcription factors such as AP-1, STATs, HIF-1, Egr-1, and IRF1also contribute to neuroinflammation (Kwon et. al. 2004; Kim et al. 2002; Wang et al. 2008; Zhang et al. 2006; Friedle et al. 2011; Iadecola et al. 1999). In contrast, transcription factors such as PPARs or Nrf2 play an anti-inflammatory role in glial activation (Drew et al. 2006; Lee et al. 2005; Shih et al. 2003). In addition, CCAAT/enhancer binding protein β (C/EBPβ), a group of the C/EBP subfamily of bZIP transcription factors also contributes to proinflammatory gene expression during glial cell activation. At least three N-terminally truncated isoforms of C/EBPβ are known to occur in various tissues (Ossipow et al. 1993; Welm et al. 1999). C/EBPβ not only plays an important role in inflammation, but also functions in modulation of cell energy metabolism, cell proliferation and differentiation (Ramji and Foka 2002). It is becoming increasingly evident that promoters of many pro-inflammatory genes contain putative C/EBPβ consensus sequences (Reddy et al. 2000) and C/EBPβ levels are upregulated in response to pro-inflammatory stimuli in macrophages (Bradley et al. 2003) and glial cells (Cardinaux et al. 2000). It is interesting to note that C/EBPβ deficiency provides neuroprotection following ischemic (Kapadia et al. 2006) and excitotoxic injuries (Cortes-Canteli et al. 2008) .
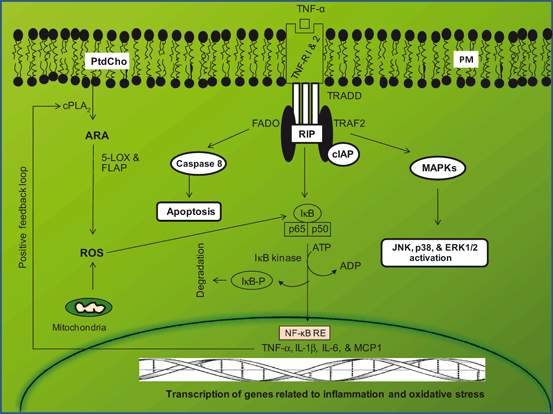
Fig. 4.5
Contribution of tumor necrosis factor-alpha receptor-mediated signaling (TNF-R) in neuroinflammation. Phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); 5-lipoxygenase (5-LOX); reactive oxygen species (ROS); 5-LOX activating protein (FLAP); tumor necrosis factor-alpha receptor-1 and tumor necrosis factor-alpha receptor-2 (TNF-R 1 and TNF-2); TNF receptor 1-associated protein (TRADD); tumor necrosis factor receptor-associated factor adaptor protein 6 (TRAF6); receptor-interacting protein (RIP); inhibitor of apoptosis protein (cIAP1); MEK kinase (MEKK); c-Jun N-terminal kinase (JNK); NF-kappaB (NF-kB); NF-kappaB response element (NF-kB-RE); inhibitory subunit of NF-κB (I-κB); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukine-6 (IL-6); monocyte chemotactic protein-1 (MCP1)
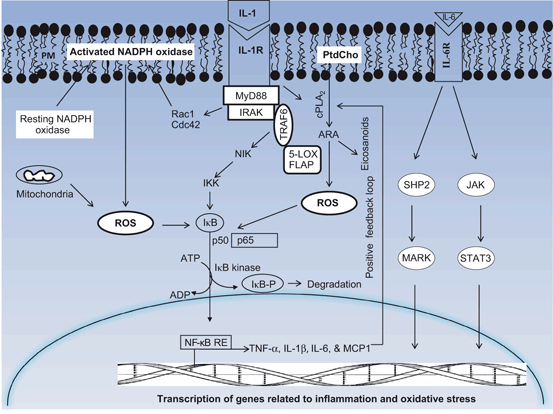
Fig. 4.6
Contribution of interleukin-1 and interleukin-6 receptor-mediated signaling (IL-1R and IL-6R) in neuroinflammation. Plasma membrane (PM); phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); adaptor protein (MyD88); 5-lipoxygenase (5-LOX); 5-LOX activating protein (FLAP); reactive oxygen species (ROS); tumor necrosis factor receptor-associated factor adaptor protein 6 (TRAF6); NF-κB-inducing kinase (NIK); IκB kinase (IKK); NF-kappaB (NF-kB); NF-kappaB response element (NF-kB-RE); inhibitory subunit of NF-κB (I-κB); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukine-6 (IL-6)
4.4 Contribution of Cytokine and Chemokine Receptors in Neuroinflammation
Cytokines and chemokines are proteins and peptides produced and released by different cells, for example: leukocytes, muscle cells, and neurons . These proteins act in a pleiotropic way or in synergy with other substances and can modulate the production of other cytokines. Structural studies have allowed the classification of these proteins into different structural classes such as the helical cytokines, the trimeric tumor necrosis factor (TNF) family, the cysteine knot growth factors and the β-trefoil growth factors (Farooqui and Horrocks 2007). Cytokines can also be classified according to the type of receptor they bind, which comprise six major families: class I cytokine receptors (the largest family), class II cytokine receptors, TNF receptors, tyrosine kinase receptors, and chemokine receptors (Tansey and Wyss-Coray 2008). In addition, cytokine families can also be named differently according to other aspects such as the sharing of a receptor subunit (i.e. the gp130 family) or its physiological roles (i.e. neuropoietic family, for its effects on hematopoietic and nervous system).
In brain tissue, cytokines (TNF-α, IL-1β, and IFN-γ) and chemokines (MCP1and CXCL3) mediate cellular intercommunication through autocrine (self), paracrine (local) or endocrine (distant) mechanisms (Tansey and Wyss-Coray 2008). Their actions involve a complex network linked to feedback loops and cascades. Cytokines and chemokines mediate their effects by binding to specific membrane-associated receptors that are composed of an extracellular ligand-binding region, a membrane-spanning region, and an intracellular region that is activated by binding of cytokines, and chemokines that facilitate the delivery of a signal to the nucleus (Rothwell and Relton 1993). Cytokine and chemokine-mediated signal transduction processes are extremely complex . These processes depend on several factors, such as the target cell type and state of activation, the local cytokine concentration, receptor type and interaction with other cytokine. Due to these factors, the precise effect of cytokines on neurons, astrocytes, oligodendrocytes, and microglial cells is difficult to predict. However, many studies have indicated that the magnitude and persistence elevations in cytokine and chemokine levels may be related to the intensity of neuroinflammation (Farooqui et al. 2007; Farooqui 2010a). Aging is accompanied by enhanced basal expression of proinflammatory cytokines and these same proinflammatory mediators often are associated with neural injury-related activation of microglia and astroglia . Taken together, the aging, inflammation and glial activation phenotypes serve as the basis for the “inflam-aging” hypothesis (Salvioli et al. 2006; Franceschi et al. 2007; Giunta 2008). According to this hypothesis, increased inflammation during the aging process results from dysregulation of the immune system and a progressive inability to properly handle pathological stimuli (Giunta 2008). Studies on aging confirm that proinflammatory cytokines in the IL-6 and TNF-α families are participants in the complex relationship between aging and chronic morbidity (Giunta 2008). Through JAK2/STAT3 signaling, these cytokines activate target genes involved in immune responses, differentiation, survival, apoptosis, and proliferation. Activation of the JAK2/STAT3 pathway is not only associated with trauma, but also with stroke (Yamashita et al. 2005; Xie et al. 2007; Shyu et al. 2008). Accumulating evidence suggests that aging may affect the proinflammatory and astrocytic response to stroke .
4.4.1 Contribution of TNF-α and TNF-α Receptors in Neuroinflammation
TNF-α is a pleiotropic cytokine, which is produced by microglia, astrocytes, and neurons. It is not only involved in BBB abnormality and inflammatory changes, but also contributes to vascular changes associated with brain injury (Barone et al. 1997) . TNF-α is synthesized as a 26 kDa membrane-bound polypeptide precursor that undergoes proteolysis to release a 17 kDa subunit. The proteolysis is catalyzed by TNF-α converting enzyme (TACE), a proteinase that belongs to the family of mammalian adamalysins (Moro et al. 2003). TNF-α signaling is associated with several important functions within the brain including regulation of microglial activation (Merrill 1991), modulation of glutamatergic transmission (Pickering et al. 2005) and control of synaptic strength (Beattie et al. 2002). Effect of TNF-α is mediated via two receptors, TNF-R1 (CD120a; p55/60) and TNF-R2 (CD120b; p75/80), which are located on the neural cell surface (Wajant et al. 2003). In brain, TNF-R1 is expressed in microglial cells, astrocytes and neurons. TNF-R1 is activated by both membrane-bound and soluble forms of TNF-α. The TNF-R2 is expressed primarily on hemopoietic and endothelial cells, responds to the membrane-bound form of TNF-α, and mediates limited biological responses (Wajant et al. 2003). TNF-R1 is constitutively expressed in most tissues, whereas expression of TNF-R2 is highly regulated and is typically found in cells of the immune system. Generally, the importance of TNF-R2 is likely to be underestimated, because this receptor can only be fully activated by membrane-integrated TNF, but not by soluble TNF (McCoy and Tansey 2008; Naude et al. 2011). TNF-R1 contains a cytoplasmic death domain and may directly cause apoptotic cell death. The TNF-R1transduction pathway is very complex. Receptor activation occurs by oligomerization and requires internalization of the ligand-receptor complex (Schneider-Brachert et al. 2004). TNF-R1 contains tyrosine residues in its intracellular domain . This allows for the ligand-TNF-R1 complex to be rapidly internalized by clathrin-coated pits following receptor activation, which is critical for mediating the death signaling. Within minutes of internalization, the TNF receptosome recruits TNF receptor-associated death domain (TRADD), which recruits additional proteins: receptor-interacting protein (RIP), TNF receptor-associated factor 2 (TRAF2) and FAS-associated death domain (FADD) (Fig. 4.5) . The latter two proteins recruit key enzymes that are closely associated with TNF-R1 signaling pathways (Kraft et al. 2009). For instance, caspase-8 is recruited by FADD to the TNF-R1 complex and initiates a protease cascade that leads to apoptosis. On the other hand TRAF2 recruits cellular inhibitor of apoptosis protein-1 and 2 (cIAP-1 and cIAP-2), two anti-apoptosis proteins that also have ubiquitin protein ligase activity. Moreover, TRAF2 may activate a mitogen-activated protein kinase (MAPK) pathway leading to the activation of c-Jun N-terminal kinase (JNK) that phosphorylates c-Jun, increasing its transcriptional activity. Finally, the protein kinase RIP is critical to the activation of the transcription factor NF-κB (Figiel 2008) . Therefore TNF-α binding toTNF-R1 may either contribute to apoptotic cell death or transcriptional activity (Celec 2004). NF-κB is a pivotal transcriptional factor down-stream of MAPK and PKC pathways and its activation is essential for controlling the expression of several genes involved in inflammation and cell proliferation (Celec 2004; Figiel 2008). Increase in TNF-α level has been observed in brain tissue, plasma and cerebrospinal fluid in stroke, Alzheimer disease, multiple sclerosis and Parkinson disease patients (Liu et al. 1994; Chao et al. 1994; Mogi et al. 1994; Sharief et al. 1993). Collective evidence suggests that TNF-R1 contains a death domain and activation normally leads to cellular apoptosis; however, under specific conditions, receptor activation can also lead to the activation of NFκB and contribute to cell survival through upregulation of anti-apoptotic genes including the Bcl-2 and XIAP families, and FLIP (Barkett and Gilmore 1999; Karin and Lin 2002) .
The role of TNF-R2 in the brain is not fully understood. However, it is becoming increasingly evident that TNF-R2 not only protects neurons against excitotoxic insults in vitro (Marchetti et al. 2004; Dolga et al. 2008), but also promotes neuronal survival as well as oligodendrocyte regeneration after ischemic and neurotoxic insults, respectively. Several pathways including PKB/Akt-dependent activation of NFκB mediate the expression of anti-apoptotic and/or neurotrophic factors (Marchetti et al. 2004; Yabe et al. 2001). In addition, PKB/Akt signaling directly exerts anti-apoptotic effects (Yabe et al. 2001) and is involved in mediating cell protection in neurodegenerative diseases (Noshita et al. 2002; Gary and Mattson 2001; Yano et al. 2001). Based on these observations, it is suggested that TNF-R2 is linked with cell growth and survival. However, it can cooperate with theTNF-R1 and contribute to neural cell death. Thus, while an elevation in TNFα in the brain is often considered an indicator of microglia activation and neuroinflammation, a number of other factors come into play to determine the final outcome .
4.4.2 Contribution of IL-1α and IL-1β and Their Receptors in Neuroinflammation
Interleukin-1 (IL-1) is a family of pleiotropic cytokines (17 KDa) , which mediates numerous actions in the brain, including neuroinflammation, fever, appetite, and hippocampal-dependent learning and memory. In addition to IL-1α and IL-1β, several other members of IL-1 family are known to occur in the brain. All members of IL-1 family are encoded by separate genes, which share some sequence homology. IL-1α and IL-1β produce similar biological actions in the brain. In addition to these two IL-1 receptor agonists, a native IL-1 receptor antagonist (IL-1ra) also maps to the IL-1 gene cluster on human chromosome two. All three proteins of IL-1α and IL-ra are synthesized from their precursors (pro-IL-1α and pro-IL-1ra), which possess biological activity. Pro-IL-1β, however, requires cleavage by caspase-1 (IL-1β converting enzyme, ICE) to become biologically active. IL-1α and IL-1β exert their action by interacting with IL-1 receptors (IL-1-Rs) and its accessory protein (Wesche et al. 1997). IL-1R1 is expressed throughout the rodent brain, with levels generally highest in neuronal rich areas including the dentate gyrus, the pyramidal cell layer of the hippocampus, and the hypothalamus (Farrar et al. 1987; Cunningham et al. 1993). Interactions between IL-1α and IL-β with IL-1R result in recruitment of the IL-1R accessory protein initiating a series of downstream signaling pathways that result into transcriptional changes as well as posttranslational modification of proteins. Two IL-1Rs have been identified so far: The type I IL-1R (IL-1R1) is generally accepted as the receptor through which IL-1 activates cellular signaling, whereas the type II IL-1R (IL-1R2) serves as a decoy receptor. There are also two different forms of IL-1R accessory proteins : The classical IL-1R accessory protein (IL-1RAcP) is expressed ubiquitously, whereas IL-1RAcPb, a novel isoform of IL-1RAcP due to alternative splicing of IL-1RAcP, is mainly expressed in CNS neurons (Smith et al. 2009; Huang et al. 2011). The IL-1R1/IL-1RAcP receptor complex is thought to mediate most, if not all, IL-1–induced effects including its involvement in ischemia-induced brain damage, activation of JNK signaling pathway, and repression of IFN-γ mRNA expression (Loscher et al. 2003; Tonzani et al. 2002). In neurons, IL-1 induces its signaling pathways through classical NF-κB or p38 MAPK pathway, such as Src and Akt pathways (Sanchez-Alavez et al. 2006; Davis et al. 2006) (Fig. 4.6) .
IL-1β is a pro-inflammatory cytokine, which is associated with several neuro-immunological and neurophysiological activities in the brain. As stated earlier, IL-1α and IL-1β have 25 % of sequence similarity at the protein level. Pro-IL-1β is synthesized in the cytosol of activated cells without a signal sequence, thus precluding secretion via the classical ER-Golgi route (Rubartelli et al. 1990). Processing of pro-IL-1β to its active form requires caspase-1 (Thornberry et al. 1992), itself activated by a molecular scaffold termed the inflammasome (Martinon et al. 2002). The binding of IL-1β with IL-1β receptors results in the induction of several inflammatory cytokines, such as IL-6 and TNF-α along with increased expression of PLA2 , cyclooxygenase-2 (COX2) and inducible NO synthase (Ballou et al. 1992). These enzymes produce ARA , prostaglandins, and nitric oxide, respectively .
Like IL-1α, IL-1β interacts with the ligand-binding chain of IL-1β receptor (IL-βR). This is followed by recruitment of the coreceptor chain, termed the receptor accessory protein (IL-1RAcP) (Dunne and O’Neill 2003). The intracellular signal transduction is initiated with recruitment of the adaptor proteins MyD88 and Toll-IL-1 receptor domain followed by recruiting IL-1 receptor-associated kinase (IRAK) to this complex (Akira and Takeda 2004; Dinarello 2011). IRAK is phosphorylated at the receptor complexes and then in turn brings tumor necrosis factor-α receptor-associated factor 6 (TRAF6) to transforming growth factor β activated kinase (TAK1). Activated TAK1 subsequently triggers a number of downstream signaling cascades, including NF-κB, p38-MAPK, and JNK, leading to the activation of transcription factors such as NF-κB and AP-1 (Bird et al. 1994; Ninomiya-Tsuji et al. 1999; O’Neil 2000; Wang et al. 2001; Li and Qin 2005).Collective evidence suggests that TNF-α and IL-1β trigger biologically indistinguishable effects by activating the same set of transcription factors, the two cytokines are structurally unrelated polypeptide that mediate their effects through distinct and structurally unrelated cell-surface receptors (Fig. 4.5 and 4.6) .
4.4.3 Contribution of IL-6 and IL-6 Receptors in Neuroinflammation
The Interleukin-6 (IL-6) is a prototypical four-helix bundle cytokine that plays an important role in regulating the pro-inflammatory and anti-inflammatory responses (Moldoveanu et al. 2001). However, due to its ability to stimulate the hypothalamus-pituitary-adrenal axis to produce cortisol and anti-inflammatory cytokines, such as interleukin-4 (IL-4), IL-6 may produce anti-inflammatory effect (Moldoveanu et al. 2001). In brain, IL-6 mediates both neurodegenerative (Morales et al. 2010) and neuroprotective effects (Peng et al. 2005; Godbout and Johnson 2004). IL-6 induces its biological effects through a hexameric receptor ligand complex including the gp130 receptor and the IL-6 receptor (Ward et al. 1994). Distinct regions of gp130 activate specific signal transduction pathways, such as the Janus kinase (JAK) signal transducer and activator of transcription (STAT), mitogen-activated protein kinase (MAPK)/cAMP responsive element-binding protein (CREB), Ras-MAPK, and PtdIns-3 kinase (Islam et al. 2009). These pathways are not only closely associated with the development of neuroplasticity, neurogenesis and neuronal differentiation, but also involve cAMP response element binding (cAMP)-mediated progenitor proliferation, and neuronal survival (Islam et al. 2009). In addition, (CREB), Ras-MAPK, and PtdIns-3 kinase also contribute to the enhancement in LTP, and memory consolidation (Vitkovic et al. 2000; Viviani et al. 2007). Through the involvement and activation of these pathways, IL-6 exerts neuroprotective and neuroproliferative effects. IL-6 also regulates survival of differentiated neurons and the development of astrocytes (Murphy et al. 1997). Some in-vitro studies have indicated that the release of IL-6 by activated microglia inhibits neurogenesis by approximately 50 % while other studies indicate that IL-6 promotes the differentiation of neural stem cells (NSCs) (Monje et al. 2003; Oh et al. 2010) . It is also reported that IL-6 induces NSC proliferation via the JAK2/STAT3 and MAPK pathways (Kang and Kang 2008; Bowen et al. 2011). Collective evidence suggests that IL-6 plays an important role in neuroproliferation, neurogenesis and memory formation .
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


