Fig. 8.1
Hyperglycemia-mediated formation of superoxide in the mitochondrial electron chain. Manganese-dependent superoxide dismutase (MnSOD); copper/zinc-dependent superoxide dismutase (CuZnSOD); hydrogen peroxide (H2O2); and superoxide (O2 −)
Thus, hyperglycemia-mediated generation of O2 •− inhibits glyceraldehyde 3-phosphate dehydrogenase (GAPDH) activity in vivo by modifying the enzyme with polymers of ADP-ribose (Fig. 8.2) (Du et al. 2003). Specific inhibitors of poly(ADP-ribose) polymerase (PARP) prevent the inhibition of GAPDH. Under normal conditions, PARP resides in the nucleus in an inactive form. Under hyperglycemic conditions DNA damage due to increase in ROS production results in activation of PARP in the nucleus. PARP splits the NAD + molecule into nicotinic acid and ADP-ribose. PARP then proceeds to make polymers of ADP-ribose, which accumulate on GAPDH and on other nuclear proteins. GAPDH is commonly thought to reside exclusively in the cytosol. However, it normally shuttles in and out of the nucleus, where it plays a critical role in DNA repair (Sawa et al. 1997; Du et al. 2003). Under physiological conditions, glucose is metabolized to glyceraldehyde-3-phosphate (G3P) via glycolysis. G3P is then converted to 1,3-diphosphoglycerate by GAPDH, which is upregulated and G3P is further metabolized to form pyruvate. Under hyperglycemic conditions, insulin resistance produces down regulation of GAPDH not only slowing glucose metabolism but increasing glucose metabolism via the polyol pathway (Phillips and Thornalley 1993; Beisswenger et al. 2003; Alexander et al. 1988) involving the enzyme aldose reductase. This enzyme utilizes a wide variety of carbonyl compounds as substrates and reduces them into sugar alcohols (polyols). It uses the nicotinic acid adenine dinucleotide phosphate (NADPH) as a cofactor. NADPH is the essential cofactor for regenerating a critical intracellular antioxidant (reduced glutathione). By reducing the amount of reduced glutathione, the polyol pathway increases susceptibility to intracellular oxidative stress. Accumulating evidence suggests that in polyol pathway, glucose is transformed into sorbitol by the enzyme aldose reductase and sorbitol, which is then oxidized to fructose by the enzyme sorbitol dehydrogenase (SDH) with NAD + as a cofactor (Giacco and Brownlee 2010). The production of sorbitol from glucose increases osmotic stress, which is associated with the pathophysiology of diabetic complications (Fig. 8.2). Accumulating evidence suggests that there are three potential mechanisms by which the polyol pathway contributes to oxidative stress. First, under hyperglycemic conditions, about 30 % of the glucose is channeled into aldolase reductase-dependent polyol pathway, which depletes NADPH and consequently reduces levels of GSH (Cheng and Gonzalez 1986). Second, oxidative stress is generated during the conversion of sorbitol into fructose by sorbitol dehydrogenase. In this step, the co-factor NAD + is transformed into NADH by sorbitol dehydrogenase. NADH is a substrate for NADH oxidase, which facilitates the formation of superoxide anions (Morre et al. 2000). Third, the polyol pathway converts glucose into fructose, and fructose can be further metabolized into fructose-3-phosphate and 3-deoxyglucosone, which are much more potent non-enzymic glycation agent than glucose (Hamada et al. 1996a, 1996b). Thus, the utilization of glucose through the polyol pathway increases the formation of advanced glycation end-products (AGEs) , ultimately leading to ROS generation. Furthermore, aldose reductase has broad substrate specificity. It recognizes short chain sugar derived carbonyls (glycoaldehyde, methylglyoxal) , medium chain hydrophobic aldehydes (4-hydroxynonenal, hexenal) and phospholipid aldehydes (1-palmitoyl-2-oxo valeroyl phosphatidylcholine, POVPC). Aldose reductase also catalyzes the reduction of the glutathione conjugates of 4-HNE and acrolein. Based on this substrate specificity, it is proposed that the enzyme is involved in the metabolism and detoxification of AGEs and advanced lipoxidation end products (ALEs) precursors and therefore it retards tissue injury and inflammation associated with ALEs and AGEs accumulation. Pharmacological inhibition of the aldose reductase exerts significant benefit against the development of various diabetic complications in animal models (Altan 2003; Drel et al. 2008). Downregulation of the polyol pathway activity by inhibiting aldose reductase prevents high-glucose-induced diacylglycerol accumulation and PKC activation in smooth muscle cells (Ramana et al. 2005, 2006). Inhibition of aldose reductase also retards high-glucose-induced stimulation of the extracellular signal-related kinase/mitogen-activated protein kinase and phosphatidylinositol 3-kinase (Campbell and Trimble 2005) and activation of NF-κB (Ramana et al. 2004), thereby decreasing smooth muscle cells chemotaxis, vascular inflammation, and adhesion. Similar processes may occur in neurons and glial cells under hyperglycemic conditions.
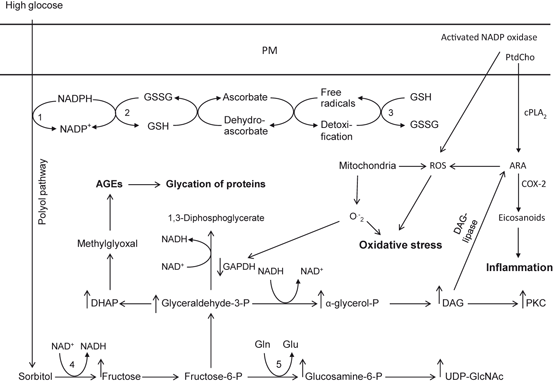
Fig. 8.2
Glucose metabolism during hyperglycemia is associated with Polyol, hexosamine, protein kinase and advanced glycation end products pathways, Plasma membrane (PM); Aldolase reductase (1); glutathione reductase (GDR) (2); glutathione peroxidase (GPx) (3); sorbitol dehydrogenase (4); glutamine fructose-6 phosphate amidotransferase (5); phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); cyclooxygenase-2 (COX-2); diacylglycerol (DAG); reactive oxygen species (ROS); advanced glycation products (AGEs); reduced glutathione (GSH); oxidized glutathione (GSSG); glyceraldehyde 3-phosphate dehydrogenase (GAPDH); reduced nicotinamide adenosine dinucleotide phosphate (NADPH); and oxidized form of nicotinamide adenine dinucleotide (NAD+)
As stated above, hyperglycemia also results in production of AGEs, which are formed by the nonenzymatic reaction of glucose and glucose-derived compounds with proteins. The AGEs are formed when α-dicarbonyl and oxoaldehydes (4-HNE and acrolein) react with free amines or amino groups of proteins to form a labile Schiff base (Maillard reaction). A cascade of reactions results thereafter in the formation of AGEs, which are composed of irreversibly cross-linked heterogeneous protein aggregates.
Methylglyoxal is generated nonenzymatically from the oxidation and spontaneous dismutation of intermediates in the glycolysis pathway or enzymic oxidation reaction catalyzed by peroxidases, whereas other AGE precursors such as deoxyglucosone are generated from fructose or from nonenzymatic degradation of Amadori rearrangement compounds (Fig. 8.3) (Conklin et al. 2007). Increase in plasma levels of methylglyoxal significantly correlates with the degree of hypertension in spontaneously hypertensive rats (Wang et al. 2005) as well as in patients with type 2 diabetes (Wang et al. 2007). This metabolite reduces activity of antioxidant enzymes like glutathione reductase and glutathione peroxidase (Wu and Juurlink 2002), leading to increased oxidative stress, which in turn mediates the pathophysiological changes in diabetes, hypertension, and aging (Ceriello and Motz 2004). Hypertension is a multifactorial condition cause by an increase in renin angiotensin aldosterone system (RAAS) activity, increase in uric acid, insulin resistance, renal disease and oxidative stress (Ceriello 2008; Manrique et al. 2009; Dhar et al. 2013). The RAAS plays an important role in maintaining fluid balance, vascular tone and blood pressure (Fyhrquist and Saijonmaa 2008; Zaman et al. 2002). Elevation in angiotensin II levels increases oxidative stress (Manrique et al. 2009; Hitomi et al. 2007) due to interactions of Angiotensin II with type I receptor (AT1 receptor). These interactions results in potent vasoconstriction, proinflammatory, pro-oxidative, proliferative and hypertrophic effects.
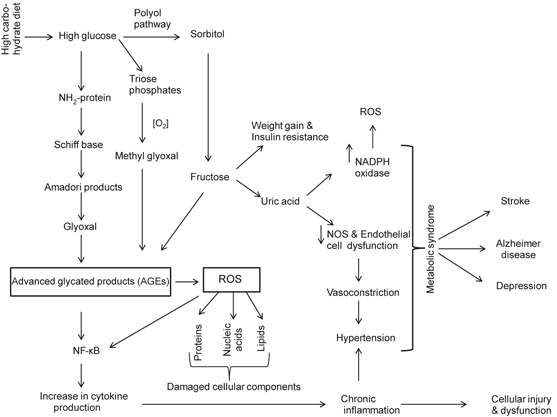
Fig. 8.3
Neurochemical effects of high carbohydrate diet on the brain. Reactive oxygen species (ROS); advanced glycation products (AGEs); and nuclear factor-kappaB (NF-κB)
Methylglyoxal is a major precursor for the formation of advanced glycation end products (AGEs) (Desai and Wu 2007). AGEs are generated normally as tissues age, but they are formed at an accelerated rate during diabetes (Wautier and Schmidt 2004). Production of AGE precursors damages cells by three general mechanisms including (a) modification of intracellular proteins by AGEs results in alterations in function, such as resistance of glycated proteins to lysosomal enzyme degradation and poor recognition of glycated haemoglobin by lipoprotein receptors and scavenger receptors (Zimmermann et al. 2001), (b) modification of extracellular matrix components by AGE precursors produces abnormalities in between other matrix components and matrix integrins receptors, which are located on the surface of cells, and (c) modification of plasma proteins by AGEs. AGEs interact with AGE receptors (RAGE), which are found on macrophages, vascular endothelial cells, vascular smooth muscle cells, neurons, astrocytes and microglial cells to induce the production of ROS, which in turn activates the pleiotropic transcription factor NF-κB, triggering multiple pathological changes in gene expression involved in proinflammatory events (Fig. 8.4) (Lue et al. 2001; Sasaki et al. 2001; Goldin et al. 2006). RAGEs modulate many signal transductions pathways associated not only with generation of more oxidative stress, but also inflammatory events (Fig. 8.5). In addition, hyperglycemia-mediated insulin resistance activates protein kinase C isoforms (due to increase in intracellular levels of diacylglycerol) and downregulates endothelial NO synthesis, as well as a induces a variety of other deleterious vascular effects, such as coagulation abnormalities, increase in production of vasoconstrictors (e.g. endothelin) and enhancement in vascular permeability along with pathological alterations in angiogenetic pathways (Sheetz and King 2002; Coppey et al. 2003). Finally, another pathway for hyperglycemia-mediated effects involves fructose-6 phosphate. This metabolite is converted by the enzyme glutamine:fructose-6 phosphate amidotransferase into UDP–N-acetylglucosamine (UDP-GlcNAc) leading to over-modification of various proteins with N-acetyl-hexosamine (Brownlee 2005) (Fig. 8.2). Collective evidence suggests that hyperglycemia-mediated increase in ROS production causes a cascade of cellular metabolic alterations, such as increase in polyol pathway influx, generation of AGEs, activation of NF-κB along with increase in hexosamine pathway (Brownlee 2005). These downstream effects may lead to blood-flow abnormalities, increased vascular permeability, angiogenesis, capillary occlusion, and pro-inflammatory gene expression (Koya et al. 1997). In addition, lipotoxicity, sympathetic nervous system activation, and extracellular matrix deposition also contribute to tissue damage caused by hyperglycemia (Dungan et al. 2009). Thus, hyperglycemia-mediated insulin resistance (Hoehn et al. 2009) is a key component of various health problems caused by the prolonged consumption of high carbohydrate diet resulting in positive energy imbalance (overeating) along with lack of physical activity. These processes result in the development of oxidative stress and low grade inflammation. The major health problems caused by hyperglycemia and insulin resistance include metabolic syndrome (MetS) , heart disease, cerebrovascular diseases, Alzheimer disease, and aging (Fig. 8.3) (Moreira et al. 2007; Ning et al. 2011) .
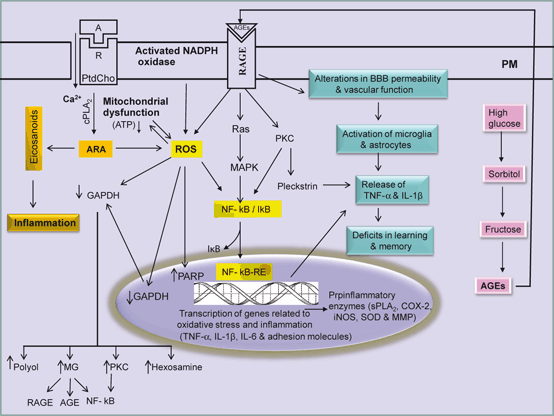
Fig. 8.4
Generation of AGE during hyperglycemia and interactions of AGEs with RAGE and RAGE-mediated signal transduction processes in the brain. Agonist (A); receptor (R); plasma membrane (PM); phosphatidylcholine (PtdCho); arachidonic acid (ARA); cytosolic phospholipase A2 (cPLA2); reactive oxygen species (ROS); nuclear factor-kappa B (NF-κB); nuclear factor-kappa B response element (NF-κB-RE); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukin-6 (IL-6); advanced glycation end-product (AGE); receptors for advanced glycation end-product (RAGE); and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
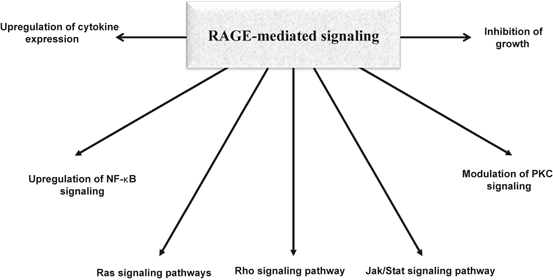
Fig. 8.5
Signal transduction pathways associated with RAGE-mediated signaling
8.5 Induction of Oxidative Stress by High Fructose Consumption
Consumption of fructose containing diet (60 % fructose) for 4 or 6 weeks not only increases serum insulin, triacylglycerol, total cholesterol, free fatty acids, uric acid , leptin, and lipid peroxide levels in rats, but also reduces serum high-density lipoprotein cholesterol and adiponectin levels. After 6-weeks of feeding fructose containing diet an increase in serum tumor necrosis factor-α and hepatic lipid peroxide levels and depletion of hepatic reduced glutathione levels is also observed (Kitagawa et al. 2012). Daily intraperitoneal administration of melatonin (1 or 10 mg/kg body weight), starting at 4-weeks high fructose containing diet feeding, attenuates above mentioned changes at 6-weeks of feeding fructose containing diet more effectively at its higher dose than at its lower dose (Kitagawa et al. 2012). Like glucose, fructose also forms AGEs. Fructose-derived AGEs not only facilitate the formation of cross-linkages between key proteins, but also interact with RAGE on the cell surfaces to induce abnormal intracellular signaling and disruption of cell function (Fig. 8.3) (Suarez et al. 1989; Seneff et al. 2011). Unlike glucose, which is utilized by all organs, fructose after absorption is solely metabolized in the liver. Fructokinase, the enzyme, which phosphorylates fructose is not regulated by negative feedback as phosphofructokinase; therefore all fructose that enters the liver cells is rapidly phosphorylated by ATP leading to ATP depletion. The depletion of ATP results in activation of AMP deaminase-1, which degrades adenine nucleotides to uric acid via xanthine oxidoreductase with the development of hyperuricemia (Nakagawa et al. 2005; Nakagawa et al. 2006; Johnson et al. 2009). Thus, uric acid is synthesized in the liver from purine compounds provided by the diet or by the endogenous pathway of purine synthesis de novo. Biologically, uric acid plays an important role in worsening of insulin resistance in animal models of diabetes and metabolic syndrome by inhibiting the bioavailability of nitric oxide, which is essential for insulin-stimulated glucose uptake (Khosla et al. 2005). Serum uric acid levels are also associated with its deleterious effects on endothelial function, platelet adhesion and aggregation, or oxidative metabolism leading to hypertension and cardiovascular disease (Alper et al. 2005). In addition, increase in uric acid promotes elevation in circulating levels of systemic inflammatory mediators such as monocyte chemoattractant protein-1, NF-κB, interleukin-1β, interleukin-6, and tumor necrosis factor-α, and vascular smooth muscle proliferation (Johnson et al. 2003; Kanellis et al. 2004).
In addition to increasing blood pressure, fructose-induced elevations in uric acid level also produces gout (Yoo et al. 2009). Lowering uric acid in fructose-fed rats reduces blood pressure, serum triglycerides, hyperinsulinemia, and weight gain suggesting that uric acid may contribute to the pathogenesis of metabolic syndrome in animal models (Nakagawa et al. 2006) . Collective evidence suggests that consumption of fructose and purine-rich foods raises uric acid levels and may play a role in the epidemic of metabolic syndrome that is occurring around the globe (Cirillo et al. 2006, 2009).
It is well known that uric acid is the end product of nucleic acid metabolism. Increase in uric acid levels in serum (hyperuricemia) is associated with pathophysiology of gout, cardiovascular disease, hypertension, metabolic syndrome, renal diseases, and stroke (Kutzing and Firestein 2008; Kim et al. 2010) but the link between uric acid and neurological disorders is unclear and controversial (Jin et al. 2012). It is reported that reduced serum levels of uric acid have been associated with Parkinson disease , Huntington disease, and multiple sclerosis (Annanmaki et al. 2007; Schlesinger and Schlesinger 2008).
Consumption of food-enriched in fructose (high-fructose corn syrup and table sugar) and purines depletes ATP and increases the levels of uric acid in blood, serum, and CSF (Choi et al. 2005). This increase in uric acid is due to stepwise degradation of AMP via AMP deaminase, 5-nucleotidase, purinenucleoside phosphorylase, xanthine oxidase resulting in the synthesis of uric acid (van den Berghe et al. 1977). This increase in uric acid may be responsible for hypertension. In brain, uric acid acts as an antioxidant that reduces oxidative stress on neurons. Uric acid reacts with peroxynitrite and stimulates the expression of extracellular SOD which may exert additional antioxidant protection. Uric acid produces beneficial effects in several neurological disorders, such as multiple sclerosis, Parkinson disease, and ALS. In mouse model of experimental autoimmune encephalomyelitis (EAE) and ALS, uric acid produces neuroprotective effects by inactivating peroxynitrite, preventing blood-brain barrier breakdown, and reducing neuroimmune responses (Kokic et al. 2005). It also stabilizes ascorbate, possibly by forming complexes with iron ions, and scavenges nitrogen radicals (Schwarzschild et al. 2008). Administration of uric acid reduces the exacerbation of the oxidative stress and mitochondrial dysfunction in human dopaminergic cells exposed to the pesticide rotenone or to iron ions supporting the view that uric acid may produce nauroprotective effects in cell culture model of PD (Duan et al. 2002). However, high levels of serum uric acid increase the risk of stroke and silent brain infarction (SBI) (Heo and Lee 2010). Although, the mechanism involved in uric acid-induced SBI is not known, but it is proposed that uric acid may enhance inflammatory activity, and reduce insulin transport into the brain (Fig. 8.6) (Lusis et al. 2008). Since majority of SBI are caused by small vessel occlusion, the risk factors of lacunar infarctions can be the plausible suspects of SBI in MetS. Conversely, uric acid can also function as a pro-oxidant, either by generating radicals during its degradation or by stimulating NADPH oxidase (Sautin et al. 2007). Uric acid can also stimulate innate immunity through the effects of microcrystalline uric acid on the function of dendritic cells and T cells (Shi et al. 2006).
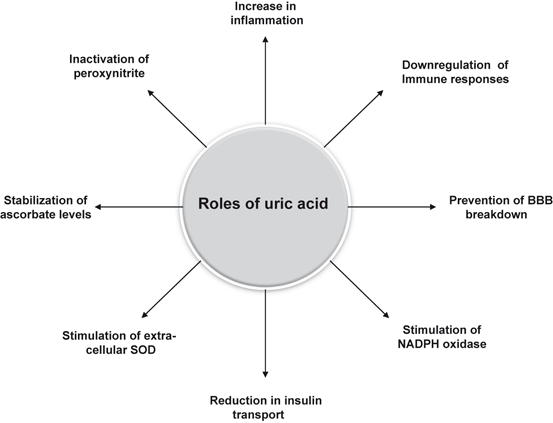
Fig. 8.6
Roles of uric acid in the brain
8.6 Contribution of ROS in Neurodegeneration
The term neurodegeneration is defined as a continuous loss of neurons often induced by protein aggregates formed of abnormally modified proteins (excessive misfolding). The accumulation of these misfolded proteins can result in a progressive loss of neurons in an age-dependent manner (Smith et al. 2002; Farooqui 2010). There is overwhelming evidence that brain tissue in patients with neurotraumatic (stroke, epilepsy, traumatic head injury, and spinal cord trauma) , neurodegenerative (Alzheimer disease, Parkinson disease , and amyotrophic lateral sclerosis) , and neuropsychiatric (depression, schizophrenia, and autism) disorders is exposed to oxidative stress during the course of disease process (Farooqui 2010). Oxidative stress in above mentioned neurological disorders is manifested through high levels of oxidized proteins, lipid peroxidation end products, and oxidative modifications in nuclear and mitochondrial DNA (Fig. 8.7) (Forster et al. 1996; Mohsen et al. 2005; Hamilton et al. 2001; Farooqui 2010; Farooqui 2011).
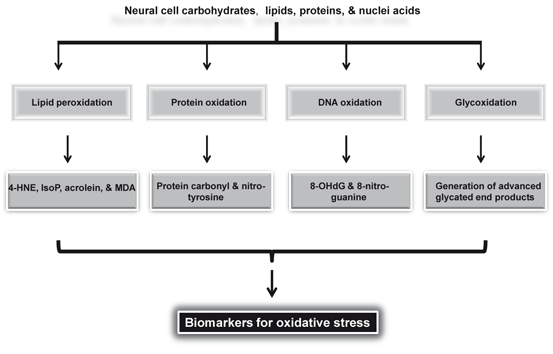
Fig. 8.7
Substrate and biomarkers for oxidative stress. 4-Hydroxynonenal (4-HNE); isoprostanes (IsoP); malondialdehyde (MDA); and 8-hydroxydeoxyguanosine (8-OHdG)
ROS mediated oxidation of protein side-chains is accompanied by the introduction of hydroxyl groups or in the generation of protein based carbonyl groups (Davies 2005), which are introduced in proteins by oxidizing amino acid residue side-chain hydroxyls into ketone or aldehyde derivatives (Davies 2005; Dalle-Donne et al. 2006) including (a) introduction of carbonyl groups in proteins by direct oxidation of lysine, arginine, proline and threonine residues, or from the cleavage of peptide bonds by the α-amidation pathway or by the oxidation of glutamyl residues; and (b) ROS can also react with other molecules, such as lipids, DNA and sugars generating reactive carbonyl derivatives and aldehydes, which may in turn react with proteins and form protein-bound carbonyls. The increase in oxidative stress in brains patients with neurotraumatic and neurodegenerative diseases is also supported by the increased brain content of iron and copper, which are capable of stimulating free radical formation (e.g. hydroxyl radicals via Fenton reaction). Measurement of protein carbonylation is a good marker for the extent of oxidative damage of proteins involved in ROS-mediated damage in neurotraumatic, neurodegenerative, neuropsychiatric diseases (Korolainen et al. 2007; Farooqui 2010). It should be noted that AGE modified proteins have a net negative charge that accumulates during their formation by glycation and oxidation (Fritz 2011; Xie et al. 2008). The second main feature is that modifications of proteins by AGEs lead to creation of multiple covalent cross-links resulting in higher molecular mass molecules (multimers). This ligand geometry has been reported to be important for the activation of RAGE (Fritz 2011).
As stated in chapter 7 that enzymic and non-enzymic oxidation of arachidonic acid results in production of ROS and formation of 4-hydroxy-2,3-nonenal (4-HNE), acrolein, malondialdehyde and F2-isoprostanes (Farooqui 2010). Levels of these lipid mediators are markedly increased in neurological disorders compared to age-matched controls (Farooqui 2011). 4-HNE not only modifies proteins, such as neuronal glucose and glutamate transporters, Na-K ATPases, and kinases, but also dysregulates intracellular calcium signaling, which ultimately lead to apoptotic neural cell death (Keller et al. 1997; Mattson and Chan 2003; Tamagno et al. 2003). In addition, 4-HNE also impairs the function of neuronal glucose transporter GLUT 3 indicating that glucose utilization can be linked with arachidonic acid metabolism (Farooqui 2011).
ROS-mediated DNA damage occurs in aged brain and brain from patients with neurodegenerative diseases, such as AD, PD, and ALS. In Haber-Weiss reaction, in the presence of free iron ions, with the participation of reactive biologically partially reduced oxygen forms (O2 − and H2O2), an •OH free radical is formed, which can react with every biological molecule being in its direct neighborhood.
Fe2+ + H2O2 → Fe3+ + OH− + •OH
Fe3+ + O2 − → Fe2+ + O2
Among ROS, the •OH is very reactive. It has a very short in vivo half-life of approximately 10−9 s and a high reactivity (Sies 1993). This makes it a very dangerous compound to the organism. Unlike superoxide, which can be detoxified by SOD, the hydroxyl radical cannot be eliminated by an enzymic reaction. As diffusion is slower than the half-life of the molecule, it reacts with any oxidizable compound in its vicinity. It can damage virtually all types of macromolecules: carbohydrates, nucleic acids, lipids, and amino acids. Thus, •OH radical particularly attacks DNA molecules (Fig. 8.8). This attack may lead to strand breaks, DNA–DNA and DNA–protein cross-linking, and formation of at least 20 modified bases adducts (Lovell and Markesbery 2007). DNA double-strand breaks may lead to genetic mutations that activate oncogenes, inactivate tumor suppressors, and change the levels or functions of ‘modifier’ proteins leading to increase in susceptibility to cancer. In addition, 4-hydroxynonenal and acrolein, which are derived from lipid peroxidation of arachidonic acid interact with DNA bases leading to the formation of bulky exocyclic adducts. Modification of DNA bases by direct interactions with hydroxyl radicals or α, β-unsaturated aldehydes can not only lead to mutations, but also cause alterations in protein synthesis. Breakdown of DNA by hydroxyl radicals results in elevations of 8-hydroxyguanine (8-OHG), 8-hydroxyadenine (8-OHA), 5-hydroxycytosine (5-OHC), and 5-hydroxyuracil (a nonenzymic breakdown product of cytosine), in both nuclear and mitochondrial DNA (mtDNA). In AD, these markers are localized in Aβ plaques and NFTs (Mecocci et al. 1994). Increased levels of DNA strand breaks have been found in AD. They were first considered to be part of apoptosis, but it is now widely accepted that oxidative damage is responsible for DNA strand breaks and this is consistent with the increased free carbonyls in the nuclei of neurons and glia in AD. The induction of heme oxygenase-1, an antioxidant enzyme involved in the conversion of heme to bilirubin, is increased in AD brains and is tightly correlated with NFTs.
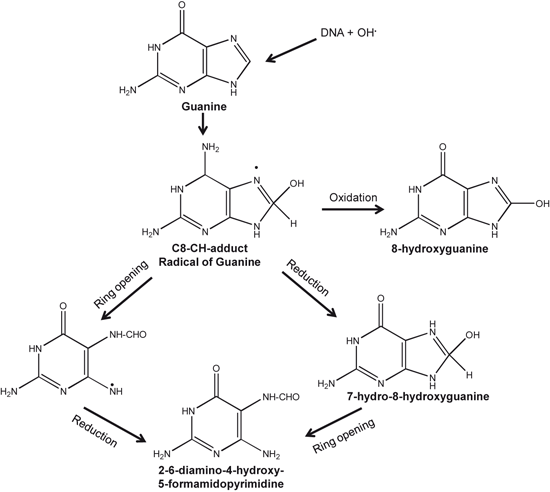
Fig. 8.8
Reactions showing attack of hydroxyl radical on guanine base of DNA
8.7 Contribution of AGEs in Neurodegeneration
Accumulation of AGEs in neural cells and tissues is a normal feature of aging, but is accelerated in hyperglycemia, diabetes, and AD. Accumulation of AGEs in the brains impairs neural cell signaling not only through direct covalent crosslinking of AGEs with various domains of its receptors, but also by interfering signal transduction processes modulated through RAGE, which is multiligand receptor (Mol mass 35 kDa) composed of three extracellular Ig-like domains (VC1, C1, C2), a single transmembrane domain, and a short cytoplasmic tail (Bierhaus et al. 2005; Takuma et al. 2009; Farooqui 2013) (Fig. 8.9). RAGE gene is located on chromosome 6 in humans between genes coding for class II and class III major histocompatibility complexes (Sugaya et al. 1994). AGEs produce their effects by binding with the VC1 domain of the RAGE receptor. AGE-RAGE interactions not only increase the phosphorylation of p21ras, the mitogen-activated protein kinases, extracellular signal-regulated kinase 1/2 and p38, but also activate GTPases Cdc42 and Rac (Fig. 8.9). These processes ultimately cause activation and translocation of NF-κB from cytoplasm to the nucleus where it starts transcribing its target set of proinflammatory genes, such as TNF-α, IL-1β, IL-6, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 (Farooqui 2013). In addition, the binding of AGEs with RAGE on endothelial cell surface also results in activation of NADPH oxidase leading to enhancement in the production of ROS (Fig. 8.9).
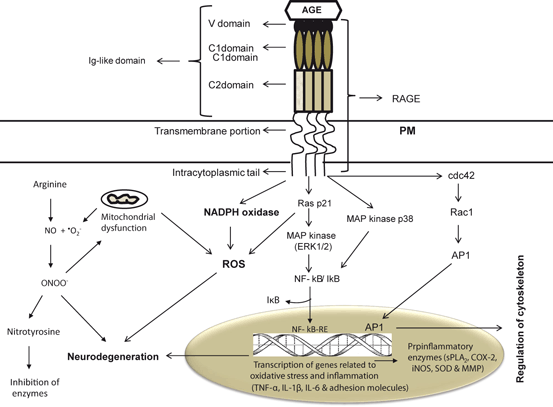
Fig. 8.9
RAGE-mediated signal transduction processes. Plasma membrane (PM); advanced glycation end products (AGEs); nitric oxide (NO); superoxide (O2); reactive oxygen species (ROS); peroxynitrite (ONOO−); nuclear factor-kappa B (NF-κB); nuclear factor-kappa B response element (NF-κB-RE); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukin-6 (IL-6); activator protein 1 (AP-1); RAS p21 protein activator 1 or RasGAP (Ras GTPase activating protein (Ras p21); and MAP kinases, p38 kinases (MAP kinase p38)
In AD, AGEs can be detected in pathological deposits such as amyloid plaques and neurofibrillary tangles. Accumulation of AGEs explains many biochemical features of AD such as extensive protein crosslinking, induction of oxidative stress, activation of astrocytic and microglial reaction and neuronal cell death. Molecular mechanisms associated with above mentioned events are not only interconnected, but are coupled with RAGE-mediated signaling at the plasma membrane of neurons, microglial cells, and endothelial cells of the vessel wall activating the transcription factor NF-κB, triggering multiple pathological changes in gene expression involved in proinflammatory events as well as neuronal expression of mitogen-activated protein (MAP) kinases signaling defects (Arancio et al. 2004) leading to neural cell death (Hadding et al. 2004). In AD, RAGE mediates transport of Aβ across the blood–brain barrier and its accumulation in the brain (Deane et al. 2003). Transgenic mice overexpressing mutant human APP and RAGE in neurons display earlier stage deficits of spatial learning/memory and more serious neuropathologic changes (Arancio et al. 2004) suggesting that AGE-RAGE interactions may facilitate neurodegeneration. RAGEs are also stimulated by beta amyloid peptide (Aβ) (Du Yan et al. 1997; Farooqui 2010). In addition, a growing body of evidence demonstrates that increase in expression of RAGE allows for more profound RAGE-induced cellular perturbation (Origlia et al. 2008). It is also reported that tau and Aβ also undergo glycation reaction (Ledesma et al. 1994; Yan et al. 2012). Glycation of Aβ has been shown to enhance its aggregation and subsequent formation of senile plaques in AD (Sasaki et al. 2001).
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Summary, Perspective and Direction for Future Studies
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Summary, Perspective and Direction for Future Studies
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


