Fig. 6.1
Low and high molecular antioxidants involved in metabolic processes associated with neural cell function
ROS perform important biological functions. Low levels of ROS are involved in signal transduction processes. H2O2, a potent oxidant freely diffuses across neural membranes to participate in intra- and intercellular signaling (Schrader and Fahimi 2006). Reversible protein oxidation by H2O2 is associated with signaling mechanism to modulate the activity of various kinases and phosphatases involved in the regulation of neural cell growth, proliferation, and apoptosis (Alder et al. 1999) Thus, serine/threonine kinases of the mitogen activated protein kinase (MAPK) family, including extracellular-regulated kinases (ERKs), c-Jun-NH2-terminal kinase (JNK), and p38 MAPK are among the signaling molecules whose activity may be regulated by ROS-mediated post-translational modification (Alder et al. 1999). ROS also regulate phosphoinositol 3 kinase (PtdIns 3K)-Akt-p53 signaling pathway. Collective evidence suggests that low levels of ROS are needed for fundamental cellular functions, such as growth, adaptation responses, and for optimal functioning of the immune system. ROS-mediated signal transduction processes are supported by several transcription factors, such as nuclear factor kappaB (NF-κB) , activator protein-1 (AP-1) , and hypoxia-inducible factor (HIF)-1α (Valko et al. 2007) (Fig. 6.2). These transcription factors reside in cytoplasm. Production of high ROS in neural cells promotes the translocation of NF-κB from cytoplasm to the nucleus, where it interacts with NF-κB response element to facilitate the expression of proinflammatory enzymes (sPLA2, COX-2, iNOS), cytokines (TNF-α, IL-1β, IL-6, IL-12) , chemokines (MIP-1α, MCPP1), growth factors, cell cycle regulatory molecules, adhesion molecule leading to inflammation (ICAM, VCAM, and E-selectin) and antiinflammatory molecules and adhesion molecules (Fig. 6.2). The DNA binding ability of NF-κB is modulated by redox status in the cell (Nishi et al. 2002). It is also shown that redox factor protein, Ref-1 reduces cysteine 62 in NFκB in the nucleus and this reaction is required for NF-κB binding to DNA (Nishi et al. 2002). Conversely, oxidation of this residue inhibits binding to DNA (Toledano and Leonardo 1991). In addition, glutathionylation of NFκB in the presence of ROS results in a decrease in its DNA binding ability and downstream transcriptional activity (Pineda-Molina et al. 2001).
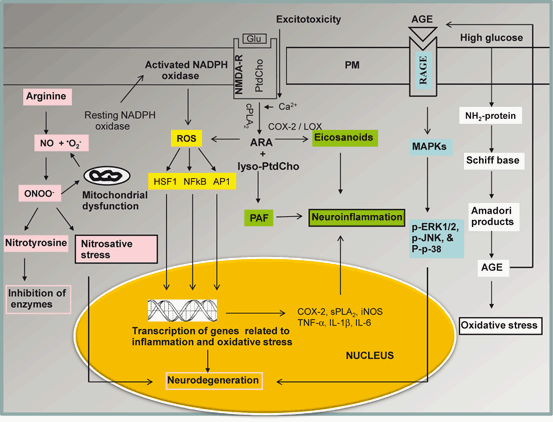
Fig. 6.2
Diagram showing the contribution of transcription factors in oxidative stress. Plasma membrane (PM), phosphatidylcholine (PtdCho), arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), platelet activating factor (PAF), N-methyl-D-aspartate receptors (NMDA-R), cytosolic phospholipase A2 (cPLA2), cyclooxygenase-2 (COX-2), lipoxygenase (LOX), reactive oxygen species (ROS), nuclear factor-kappa B (NF-κB), nuclear factor-kappa B response element (NF-κB-RE), tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6), nitric oxide (NO), peroxynitrite (ONOO-), advanced glycation end-product (AGE), receptors for advanced glycation end-product (RAGE), mitogen-activated protein kinase (MAPK), extracellular signal regulated kinase (ERK), Jun amino-terminal kinases (JNK), Heat shock factor protein 1 (HSF1), activator protein 1 (AP-1)
Similarly, AP-1 is also involved in the increased expression of adhesion molecules and inflammatory cytokines. Transcription factor, HIF-1α not only activates a broad range of genes protecting cells against hypoxia, but also binds with p53 to regulate many genes including Bax (a proapoptotic member of the Bcl-2 family) (Gibson et al. 2001). Bax inactivates Bcl-2 by forming a heterodimer. The balance between levels of bcl-2 and bax can serve as an indicator of cell survival or death (Longoni et al. 1999).
Nuclear factor-erythroid 2-related factor 2 (Nrf2), a member of the cap“n”collar transcription factor family, is a master regulator of antioxidant defense genes and drug-metabolizing enzymes (Kensler et al. 2007). ROS are known not only to oxidize the cysteine residues of Keap1, but also upregulate Nrf2 through kinase activation. Subsequent phosphorylation of Nrf2 enhances Nrf2 dissociation from Kelch-like ECH-associated protein1 (Keap1) and allows migration of Nrf2 to the nucleus, where it interacts with antioxidant response elements (ARE) and facilitates the transcription of heme oxygenase-1 (HO-1), glutathione synthase (GS), NADP(H) quinine-oxidoreductase1 (NQO1), catalase and Na + -independent cystine/glutamate exchanger (Fig. 6.3) (Papaiahgari et al. 2006; Lee et al. 2003a; Lee et al. 2003b; Wang et al. 2012). Extra-cellular signal-regulated kinase (ERK), c-Jun N-terminal protein kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathways also regulate Nrf2 transcriptional activity. ERK and JNK regulate Nrf2 pathway activity positively, whereas p38 MAPK has been reported to regulate Nrf2 both positively and negatively (Naidu et al. 2009; Singh et al. 2010). ERK, JNK, and p38 MAPK act by phosphorylating N-terminal serine residues on Nrf2, which results as a response to electrophiles and oxidative stress (Huang et al. 2002). A 50 % reduction in cerebral blood flow following ischemic injury in mice results in activation of Nrf2 in neurons predominantly in cerebellar Purkinje cells and cingulate cortex (Liverman et al. 2004) supporting the view that activation of Nrf2 is closely associated with protection against neuronal injury. A variety of stimuli have been shown to induce Nrf2 activation by disrupting the Keap1-Nrf2 complex in the cytoplasm (Kaspar et al. 2009). Hydrogen peroxide and nitric oxide have been shown to oxidize several cysteine residues on Keap1, thereby forming intra- and intermolecular disulphides and inactivating Keap1 (Dhakshinamoorthy and Porter 2004). Nrf2 activation and its downstream signaling are important mediators of antioxidant signaling during exposure to low levels of ROS. Nrf2 also increases anti-inflammatory mediators, activity of the proteasome, and other transcription factors involved in mitochondrial biogenesis (Tufekci et al. 2011). Accumulating evidence suggests that activation of Nrf2 is associated with regulation of basal and inducible expression of numerous antioxidant stress genes, which play an important role in neuroprotection against oxidative stress not only in many animal models of neurotraumatic and neurodegenerative diseases, but also in human subjects.
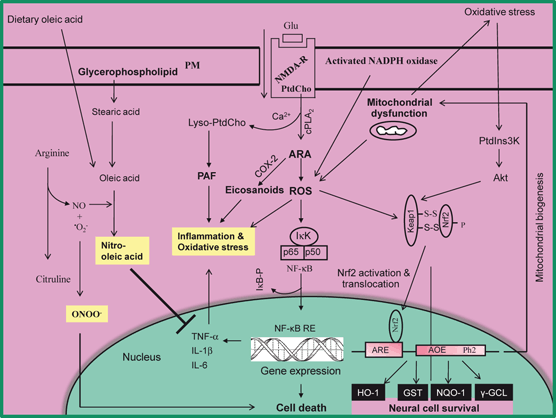
Fig. 6.3
Diagram showing the activation of Nrf2, induction of antioxidant enzymes and suppression of neuroinflammation by nitro-oleic acid following oxidative stress. Plasma membrane (PM), phosphatidylcholine (PtdCho), arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), platelet activating factor (PAF), N-methyl-D-aspartate receptors (NMDA-R), cytosolic phospholipase A2 (cPLA2), cyclooxygenase-2 (COX-2), reactive oxygen species (ROS), nuclear factor-kappa B (NF-κB), nuclear factor-kappa B response element (NF-κB-RE), tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6), nitric oxide (NO), peroxynitrite (ONOO-), phosphatidylinositide 3-kinases (PtdIns 3K), Protein Kinase B (Akt), Nuclear factor-erythroid 2-related factor 2 (Nrf2), Kelch-like ECH-associated protein 1 (Keap1), hemeoxygenase (HO-1), NAD(P)H:quinone oxidoreductase-1 (NQO1), antioxidant response element (ARE), and γ-glutamate-cysteine ligase (γ-GCL)
Under pathological conditions or after exposure to certain toxic agents greater amounts of ROS are leaked from the mitochondrial electron transport chain, possibly at the sites of Complex I (NADH ubiquinone oxidoreductase) and Complex III (ubiquinone cytochrome c oxidoreductase) (Bailey et al. 1999). High levels of ROS not only suppress protein synthesis and activate proteases, but also attack phospholipids, cholesterol, protein, and DNA to generate uniquely oxidized biomolecules that can be used as “fingerprints” to detect oxidative stress in cells (Powers and Jackson 2008) (Fig. 6.4). High levels of ROS modulate telomeres (repeats of DNA–protein complexes), the structures, which are located at the ends of chromosomes and are essential for the stability of chromosome and cell replication. Telomeres are considered as a ‘biological clock’ of the cellular ageing. Telomerase is an enzyme that adds nucleotides to telomeric DNA thereby contributing to telomere maintenance, genomic stability, functions, and proliferative capacity of the cell. Increase in intracellular ROS levels is associated with acceleration in the rate of telomere shortening. The progressive shortening of telomeres leads to senescence, apoptotic cell death, or the oncogenic transformation of somatic cells in various tissues. Telomere length is modulated by various lifestyle factors (diet, exercise, calorie restriction, alcohol abuse, and smoking). It is known to determine overall health, lifespan, and the rate at which an individual ages (Epel et al. 2004; Minamino et al. 2008; Babizhayev et al. 2010; Lin et al. 2012). Recent in vitro studies have indicated that Mediterranean diet protects the cells from oxidative stress not only by preventing cellular senescence, and cellular apoptosis, but also through the maintenance of telomere length (Boccardi et al. 2013). In addition, mitochondria also become a main target of oxidative damage because of the relatively low levels of antioxidants, such as reduced glutathione (GSH), in mitochondria compared to cytosol (Fernandez-Checa et al. 1998).
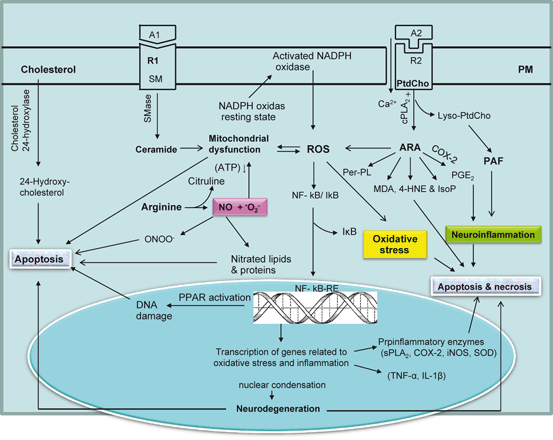
Fig. 6.4
Diagram showing contribution of phospholipids, sphingolipids, and cholesterol-derived lipid mediators in oxidative stress. Plasma membrane (PM), phosphatidylcholine (PtdCho), arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), platelet activating factor (PAF), agonist 1 (A1), receptor 1 (R1), agonist 2 (A2), receptor 2 (R2), cytosolic phospholipase A2 (cPLA2), cyclooxygenase (COX), reactive oxygen species (ROS), sphingomyelin (SM), sphingomyelinase (SMase), nuclear factor-kappa B (NF-κB), nuclear factor-kappa B response element (NF-κB-RE), tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6), nitric oxide (NO), peroxynitrite (ONOO-), secretory phospholipase A2 (sPLA2), inducible nitric oxide synthase (iNOS), superoxide dismutase (SOD), peroxidized phospholipid (Per-PL), 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), and isoprostane (IsoP)
ROS-mediated stimulation of sphingomyelinase (SMase) promotes the hydrolysis of sphingomyelin and generates ceramide, a lipid mediator that induces mitochondrial dysfunction, resulting in the production of more ROS and apoptotic cell death. Cholesterol oxides (oxysterols), which are derived from cholesterol by enzymic (cytochrome P450) and nonenzymic reactions, are markers of brain atrophy in patients with various neurodegenerative diseases (Leoni and Caccia 2011; Russell 2000; Schroepfer 2000). In brain, oxysterols have various neurochemical functions (induction of cell death, activation of inflammation, gene transcription, modulation of cognitive function, and/or oxidative processes). Therefore, oxysterols have been proposed to play major roles in the pathologies of various neurodegenerative diseases (Farooqui 2011). ROS, which are known to alter the intracellular redox balance, also facilitate the translocation of p53 to mitochondria. ROS along with 7-oxysterol and 7-ketocholesterol enhance mitochondrial translocation of Bax, mitochondrial membrane permeabilization, cytosolic release of cytochrome c, and cell death (Leoni and Caccia 2011; Russell 2000; Schroepfer 2000). In addition, 7-ketocholesterol also promotes the formation of ROS with subsequent activation of NFκB along with upregulation of interleukin-1β, interleukin (IL)-6, and IL-8 .
Direct measurement of ROS production is very difficult due to their high reactivity and low steady-state concentration. Nevertheless, the electron spin resonance (ESR) technique is a direct method of detection of species that have an unpaired electron, generally meaning that it is a free radical (Ashton et al. 1999). Common measures of bio-oxidation include the measurement of protein carbonyls as an indicator of protein oxidation; assessment of isoprostanes, malondialdehyde, and 4-hydroxyl-2-nonenol as signs of lipid peroxidation (Durand et al. 2005); and evaluation of DNA oxidation by assaying the levels of the oxidized base, 8-hydroxy-2′-deoxyguanosine (8-OH-dG) (see below) (Wink et al. 1991). The exogenous ROS burden creates an environment with oxidative stress mainly through the production of intracellular H2O2, which acts as a second messenger in cell signaling pathways. H2O2 reacts with freely accessible protein thiolate anions to form cyssulfenic acids, which is further oxidized to disulfide bonds. Most of these reactions are reversible in the presence of cellular thiol/disulfide systems such as GSH/GSSG, thioredoxin/thioredoxin reductase, and cysteine/cystine (Phillips et al. 2010). However, in the presence of excessive oxidation of cysteine and GSH depletion can lead to eventual overwhelming of endogenous oxidation-reduction (redox) reactions. Intracellular redox status is very important in protein function and to maintain cellular homeostasis. In neurotraumatic, neurodegenerative, and neuropsychiatric diseases enhanced ROS levels contribute to neuronal membrane damage not only by attacking neural cell membrane components (polyunsaturated fatty acids, sulfhydryl groups of proteins, bases of nucleic acids, and carbohydrates), but also by altering activities of various transcription factors (NF-κB, AP-1, HIF-1, and Nrf2) (Farooqui 2010). The effect of ROS-mediated damage to neural membrane phospholipids, sphingolipids, and cholesterol is cumulative and not amenable to repair, particularly in postmitotic cells such as neurons. Interactions between ROS and cellular components are known to alter cell membrane fluidity and permeability, ion transport, activities of membrane bound enzymes, protein cross-linking, inhibition of protein synthesis, and DNA damage eventually leading to apoptotic cell death (Farooqui and Horrock 2007). In addition, interactions between ROS and mitochondrial DNA (mtDNA) may lead to mutations, which may promote neuronal cell death. ROS also disrupt blood brain barrier (BBB) via matrix metalloproteinase (MMP) activation. The MMPs are zinc-containing enzymes, which are involved in the degradation of extracellular matrix around cerebral blood vessels and neurons (Gu et al. 2011). ROS-mediated activation of MMPs is involved in the stimulation of extracellular matrix degradation. This can occur at the tight junctions and therefore disrupt the vital BBB (Gu et al. 2012). It is also suggested that loss of BBB integrity due to ROS-mediated MMP activation is closely associated with cerebral ischemia – reperfusion injury (Gu et al. 2011; Gu et al. 2012).
6.3 Phospholipid-Derived Enzymic and Non-Enzymic Biomarkers of Oxidative Stress
As stated earlier, ARA is located at the sn-2 position of glycerol moiety in neural membrane phospholipids. It is released by at least two major mechanisms . A direct mechanism involves the activation of cPLA2, and an indirect mechanism, which requires the activation of phospholipase C (PLC) followed by diacylglycerol lipase (Farooqui and Horrocks 2007). The free ARA is oxidized by COXs; LOXs, and EPOXs resulting in the formation of prostaglandins (PGs) , leukotriene (LTs) , lipoxins (LXs) , and thromboxanes (TXs) , as well as hydroxyeicosatetraenoic acid (HETE) and epoxyeicosatetraenoic acids (EETs), and dihydroxyeicosatrienoic acids (DHETs) (Fig. 6.5). These metabolites are collectively known as eicosanoids. Eicosanoids produce a wide range of biological actions including potent effects on neuroinflammation, vasodilation, vasoconstriction, apoptosis and immune responses (Phillis et al. 2006). In addition to generating above mentioned metabolites, COX, LOX, and EPOX-catalyzed reactions also produce ROS. As mentioned above, ROS at low levels function as signaling molecules in the regulation of fundamental cell activities such as growth and adaptation responses, but at higher concentrations, ROS contribute to neural membrane damage when the balance between reducing and oxidizing (redox) forces shifts toward oxidative stress leading to chemical cross-linking of membrane proteins and lipids and a reduction in membrane unsaturation. The depletion of unsaturation in membrane lipids is associated with reduction in membrane fluidity, decrease in activity of membrane-bound enzymes, ion-channels, and receptors, leading to apoptotic and necrotic cell death (Farooqui et al. 2009).
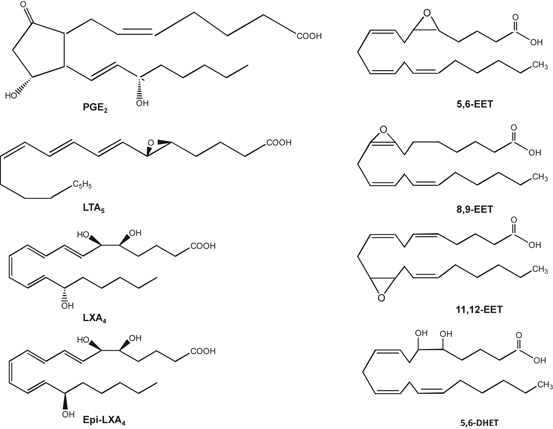
Fig. 6.5
Chemical structures of arachidonic acid-derived prostaglandins, leukotrienes, lipoxins, and epoxyeicosatetraenoic acids
Non-enzymic peroxidation of ARA results in formation of 4-hydroxynonenals (4-HNE), isoprostanes (IsoP), isoketal (IsoK) isofuran (IsoF), acrolein (Ac), and malonaldehyde (MDA) (Fig. 6.6). The first step in production of ARA-derived lipid mediators is formation of hydroperoxide (the primary product). The breakdown of hydroperoxide results in formation of secondary products, such as 4-HNE, IsoP, IsoK, IsoF, Ac, and MDA. All these products can be measured in biological fluids, but analytical procedures used are sometimes complex and require sample preparation involving extraction and purification steps (Milne et al. 2013). Non-enzymic ARA-derived lipid mediators can be divided into two categories. Mediators containing α, β-unsaturated aldehyde group, such as 4-HNE, Ac, and MDA, and lipid mediators, which contain ring structure that resemble PGs, LTs, and TXs, such as IsoP, IsoK, and IsoF. Both types of ARA-derived lipid mediators are associated with neuroinflammation and vascular dysfunction . α, β-Unsaturated aldehyde containing lipid mediators are toxic because of their high reactivity with nucleophiles and their ability to form protein and DNA adducts without prior metabolic activation (Farooqui 2011). This strong reactivity leads to electrophilic stress that disrupts normal cellular function. Furthermore, α, β-unsaturated aldehydes are reported to cause endothelial dysfunction by induction of oxidative stress, redox-sensitive mechanisms, and neuroinflammatory changes such as induction of cyclooxygenase-2 and cytokines (Farooqui 2011). It is proposed that α,β-unsaturated aldehydes act as redox signaling mediators leading to cellular and tissue injury. The maintenance of the redox balance in the vascular system is of paramount importance since increased production of ROS contributes to endothelial dysfunction and vascular disease. Oxidative stress is increasingly seen as a major upstream component in the signaling cascade involved in many cellular functions, such as neural cell proliferation, neuroinflammatory responses, and adhesion molecule stimulation (Farooqui 2011). Furthermore, α, β-unsaturated aldehydes also induce the inactivation of antioxidant enzyme such as glutathione peroxidase and thioredoxin, activation of NF-κB signaling pathway, and stimulation of inflammatory response through activation of the proinflammatory signaling pathway .
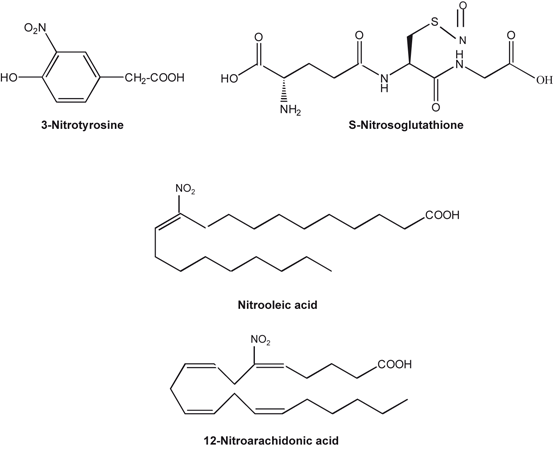
Fig. 6.6
Chemical structures of 3-nitrotyrosine, nitrosoglutathione, nitro-oleic and nitro-arachidonic acids
In contrast, non-enzymic lipid mediators of ARA produce their effects directly and via receptor-mediated mechanisms on the vasculature. For example, IsoP binds with TxA2 receptors (TPRs). These receptors are not only found in the brain, but also in macrophages or monocytes, vascular endothelial cells, and platelets. TPRs modulate antiatherosclerotic, antivasoconstrictive, and antithrombotic effects, depending on the cellular target. TPR receptors mobilize intracellular Ca2 +and are coupled with the activation of protein kinases (MAP kinase and Ca2 +/Rho kinase) (Kinsella et al. 1997; Pratico et al. 1997). F2-IsoP and its receptor also mediate their effects in vascular beds and platelet function by promoting interactions between endothelial cells and monocytes (Lahaie et al. 1998; Fam and Morrow 2003) .
Proteins are susceptible to posttranslational modifications produced by above mentioned aldehydes binding covalently to specific amino acid residues, in a process called Michael adduction. Posttranslationally modified proteins are usually removed by the proteasome. However, inhibition of the proteasome or decreasing proteasomal activity may result in an accumulation of abnormal proteins (Jung et al. 2009). In a vicious cycle these abnormal proteins either overload the proteasome resulting in blocking of its activity, or they stimulate the generation of ROS, e.g. via the activation of microglia cells. All these events may contribute to the dramatic vicious cycle: increased oxidative stress leads to modified proteins; these may inhibit proteasomal activity which contributes to an accumulation of damaged proteins and these may increase oxidative stress. It is becoming increasingly evident that protein posttranslation modifications are important mediators of brain damage because of their ability to disrupt important cellular function (Esterbauer et al. 1991; Farooqui and Horrocks 2007; Roberts et al. 2005; Farooqui 2011). Studies on oxidatively, modified proteins have shown an age-related increase in the content of protein carbonyls (Levine 2002), glycated proteins (Baynes 2001), oxidized methionine (Stdtman et al. 2005) and an accumulation of enzymes that are impaired in their catalytic activity (Ito et al. 1998). However, the best studied marker for age-related protein oxidation is currently protein carbonylation (Levine 2002) .
6.4 Carbohydrate-Derived Biomarkers for Oxidative Stress
Advanced glycation end products (AGEs) are a heterogeneous, complex group of compounds that are formed mainly via the Maillard reaction , which occurs when carbonyl group in reducing sugar reacts in a non-enzymatic way with amino acids in proteins (especially of basic lysine or arginine residues) forming a non-stable Schiff base. Further rearrangement leads to formation of a more stable ketoamine (Amadori product) (Ahmed 2005; Giacco and Brownlee 2010). Schiff bases and Amadori products are reversible reaction products. However, they can react irreversibly with amino acid residues of peptides or proteins to form protein adducts or protein crosslinks. Alternatively, they can undergo further oxidation, dehydration, polymerization and oxidative breakdown reactions to give rise to numerous other AGEs. Oxygen, reactive oxygen species (ROS) and redox active transition metals accelerate AGE formation (Ahmed 2005). AGE production not only induces impaired function, but glycated proteins become highly susceptible to oxidative damage. Glycated proteins are also resistant to degradation by lysosomal enzymes (Giacco and Brownlee 2010). AGEs play a critical role in aging, diabetes, cardiovascular diseases, and neurodegenerative diseases. Hyperglycemia is closely linked with accelerated AGEs formation (Mendez et al. 2010). The major AGEs in vivo are formed from highly reactive intermediate carbonyl groups, known as α-dicarbonyls or oxoaldehydes, including 3-deoxyglucosone, glyoxal, and methylglyoxal (MG) (Brownlee 2001; Kim et al. 2005). Levels of these aldehydes are normally kept at a low level by catabolism via the glutathione-dependent glyoxalase enzyme system. Excess MG may lead to the depletion of cysteine and glutathione, resulting in decreased antioxidant capacity (Thornalley 1993) .
6.5 Protein-Derived Biomarkers for Oxidative Stress
It is well known that proteins are the major target for oxidants (Davies 2005) . Thus, polypeptide backbone of proteins and the side chains of most amino acids are susceptible to oxidation. The non-enzymic introduction of aldehyde or ketone functional groups to specific amino acid residues constitutes the most common oxidative alteration of proteins (Bizzozero 2009). This process is called as protein carbonylation. The direct protein carbonylation can be achieved through a variety of reactions. Thus, oxidation of amino acid side chains with metals and hydrogen peroxide is known to produce semialdehyde amino acids, with the majority of these reactions occurring with lysine, arginine, and proline residues (Stadman and Berlett 1991). Alternatively, protein carbonylation can result from an indirect mechanism involving the hydroxyl radical-mediated oxidation of lipids (Schneider et al. 2001). Protein carbonylation causes mitochondrial dysfunction by inducing changes in activities of phosphate carrier protein, NADH dehydrogenase 1α subcomplexes 2 and 3, translocase of inner mitochondrial membrane 50, and valyl-tRNA synthetase (Curtis et al. 2012). Elevated protein carbonylation is also accompanied by reduction in activity of complex I, impaired respiration, increase in superoxide production, and a reduction in membrane potential without changes in mitochondrial number, area, or density indicating that protein carbonylation plays a major instigating role in mitochondrial dysfunction. Since oxidized protein is harmful to the maintenance of cellular homeostasis, it requires rapid removal by proteolytic digestion. it is shown that proteolysis is the only physiological mechanism for elimination of carbonylated proteins, as there is no evidence for enzymatic reduction of protein-bound carbonyl groups to alcohols (Bizzozero 2009). Accumulation of protein carbonyls does not only occur, but has also been implicated, in the pathophysiology of several neurological disorders, including AD (Aksenov et al. 2001), PD (Floor and Wetzel 1998), ALS (Ferrante et al. 1997), and multiple sclerosis (Bizzozero et al. 2005) .
6.6 Nucleic Acid-Derived Biomarkers for Oxidative Stress
The attack of ROS on DNA particularly hydroxyl radicals, can lead to strand breaks, DNA–DNA and DNA–protein cross-linking, and formation of at least 20 modified bases adducts (Barciszewski et al. 1999; Lovell and Markesbery 2007) . Among DNA bases, the guanine residues are most readily oxidized by the hydroxyl radical (•OH) and singlet oxygen (1O2) (Steenken and Jovanovic 1997). 8-Oxo-7,8-dihydroguanine (8-oxoG) is the most abundant ROS-related product of DNA oxidation, which has been implicated in mutagenesis (Lu and Liu 2010; Damsma and Gramer 2009). 8-Oxoguanine DNA glycosylase (OGG1) is a bifunctional DNA glycosylase that removes oxidized bases such as 8-oxo-G, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FaPyG) and 7,8-dihydro-8-oxoadenine (8-oxo-A) from the DNA (Morales-Riuz et al. 2003). This enzyme is specific for incising 8-oxoG to avoid the transversion of GC → TA, or DNA damage–induced apoptosis. It is recently shown that OGG1 can rescue neurons subjected to ischemic conditions (Liu et al. 2010). It is known that acetylation of OGG1 can increase the activity of the enzyme nearly 10-fold in a cell culture (Bhakat et al. 2006), but the presence of in vivo acetylation has yet to be reported. Many investigators have focused on oxidation of DNA (Frieberg 2003) since DNA oxidative lesions must be repaired to maintain the genomic integrity .
Oxidative damage to RNA occurs more frequently than DNA, because RNA molecules are mostly single stranded and its bases are less protected by hydrogen bonding. Moreover, most of the mRNAs are not associated with chromatin and are distributed in the cytoplasm, closer to the site, where ROS generation occurs (Radak and Boldogh 2010). There is evidence that oxidized mRNA causes errors in translation, eventually leading to the production of abnormal proteins (Tanaka et al. 2007), such as Aβ in AD, α-synuclein in PD, and mutated huntingtin in HD (Nunomura et al. 2009; Farooqui 2010). Furthermore, increase in levels of oxidatively damaged nucleic acids has been observed in numerous diseases including AD (Gackowski et al. 2008), PD (Nakabeppu et al. 2007), autoimmune diseases (Bashir et al. 1993) and cardiovascular diseases (Lee and Blair 2001). It is suggested that such damage may play an important role in the etiology of these diseases (Cooke et al. 2003; Cooke et al. 2006) .
Mitochondrial dysfunction and ROS production have also been implicated in the development of several neuropsychiatric diseases such as depression, anxiety and schizophrenia. It is becoming increasingly evident that mitochondrial dysfunction and ROS production result in severe consequences for monoamine levels in the brain, resulting in decreases in serotonin (5-hydroxytryptamine, or 5-HT), dopamine, noradrenaline, and γ aminobuteric acid (GABA) (Gardner and Boles 2010), all of which are monoamines associated with depressive, schizophrenic and anxious behaviors. For example, neurochemical links have been reported to occur between mitochondrial dysfunction and the development of schizophrenia: microscopic analysis of autopsy specimens show mitochondrial malformations and reduced density in the anterior limbic cortex and the caudate putamen nucleus, both areas highly associated with the development of schizophrenia (Ben-Shachar 2009). Mitochondrial dysfunction has also been linked to decreases in brain-derived neurotrophic factor (BDNF), decreases in hippocampal neurons and disruption of the hypothalamus-pituitary-adrenal (HPA) axis, all of which has been linked to the development of depressive and anxious symptoms (Clay et al. 2010) .
6.7 Neurochemical Aspects of Nitrosative Stress
Under physiological condition in cardiovascular and cerebrovascular systems, NO is produced in a controlled manner at low levels during the metabolism of arginine to citruline by endothelial nitric oxide synthase (eNOS) . NO is an important regulator of physiological processes in the central nervous system (CNS). It promotes optimal cerebral blood flow, consolidates memory processes, facilitates long-term potentiation, maintains sleep-wake cycles, and assists in normal olfaction (Virarkar et al. 2013). In the absence of L-arginine or co-factors, the activated eNOS does not catalyze the oxidation of L-arginine into NO. Although, the enzyme continuous with the capacity to receive and store electrons in the reductase, donating them to the oxygen substrate. Therefore, the eNOS generates superoxide instead of NO (Bahia et al. 2006). The superoxide reacts rapidly with the local NO to forms peroxynitrite (see below). This metabolite produces damage to the cellular proteins and DNA as well as induces the decoupling of eNOS, which leads to a higher production of superoxide maintaining the conditions of endothelial damage and pathophysiology of vascular diseases (Bahia et al. 2006). Arginine is alternatively metabolized by arginase to ornithine and urea. When the supply of arginine is limited, metabolism via arginase may effectively reduce production of NO (Li et al. 2001). Thus, the decrease in bioavailability of NO within the vessel wall is caused by the competitive utilization of arginine by arginase and “eNOS uncoupling.” NO enhances your blood flow when produced in low quantities by blood vessel cells. It not only helps with healthy blood pressure levels, but also supports functioning of the immune system (Alderton et al. 2001; Madigan and Zuckerbraun 2013) .
Nitrosative stress is defined as the ratio of nitrosants and antioxidants similar to oxidative stress, but with the additional involvement of reactive nitrogen species . Oxidative/nitrosative stress represents the imbalance in the production and the elimination of reactive oxygen and nitrogen species. This process is associated with the pathogenesis of neurotraumatic and neurodegenerative diseases (Farooqui 2010). These neurological conditions are accompanied by hyperactivation of the N-methyl-D-aspartate (NMDA) type of glutamate receptor, mitochondrial dysfunction, cellular aging, and accumulation of misfolded proteins, such as beta amyloid (Aβ) in Alzheimer disease (AD) , alpha-synuclein (α-Syn) in Parkinson disease (PD) , and mutated huntingtin (HT) in Huntington disease (HD) along with introduction of 3-nitrotyrosine (Fig. 6.6), S-nitrosylation of thiol and amine groups, and oxidation of thiol groups in cysteine and methionine residues (Fig. 6.6) (Pacher et al. 2007; Leon et al. 2008) . These post-translational modifications can potentially activate or inhibit target proteins, with different biological consequences. Thus, the excessive generation of NO and NO related species triggers pathological production of misfolded proteins, abnormal mitochondrial dynamics (comprised of mitochondrial fission and fusion events), and apoptotic pathways in neuronal cells (Nakamura and Lipton 2011). At high concentration NO also reacts with DNA and initiates signaling pathways that causes neural cell damage. These processes contribute to nitrosative stress in the various types of neural cells in the brain (Pacher et al. 2007; Leon et al. 2008) .
6.7.1 Nitric Oxide and its Effect on Cellular Components
NO is an important second messenger, which is synthesized by NO synthases (NOS), a family with four major types of enzymes called endothelial NO synthase (eNOS), neuronal NO synthase (nNOS), inducible NO synthase (iNOS) and mitochondrial NO synthase mNOS). Low levels of NO are produced by the eNOS whereas high levels of NO are produced by the iNOS during neuroinflammation and oxidative stress. In brain, NO not only functions as vasorelaxant, but also plays important roles in synaptic plasticity (long-term potentiation, LTP; long-term depression, LTD) and modulation of NMDA receptor (Prast and Philippu 2001; Steinert et al. 2010). NO is thermodynamically unstable molecule that reacts with other molecules, resulting in the oxidation, nitrosylation or nitration of proteins, with the concomitant effects on many cellular mechanisms. NO signaling is coupled with the activation of guanylate cyclase, modulation of MAPKs, interactions with apoptosis-related proteins, and binding with mitochondrial respiratory chain (Prest and Philippu 2001; Steinert et al. 2010). NO also plays a role in post-translational modification of proteins and protein degradation by the proteasome. Within the brain tissue, the susceptibility of neurons, astrocytes, oligodendrocytes, and microglial cells to NO exposure may be dependent on factors such as the intracellular reduced glutathione and cellular stress resistance signal pathways. Thus neurons, in contrast to glial cells, appear particularly vulnerable to the effect of nitrosative stress. Covalent reaction between NO and thiol groups of specific protein is called as S-Nitrosylation. Nitrosylation modifies function of many proteins (triose phosphate isomerase, glutamate dehydrogenase, neuropolypeptide h3, phosphoglycerate mutase1, H + -transporting ATPase, α-enolase and fructose-1,6-bisphosphate aldolase) (Reed et al. 2009). S-Nitrosylation of redox-sensitive enzyme, disulfide isomerase and dynamin-related protein 1 in the endoplasmic reticulum contribute to neurodegeneration through alterations in hydrophobicity and electrostatic properties (Uehara 2007; Nakamura and Lipton 2009; Benhar et al. 2006; He et al. 2007). The activation of NAD+ -consuming enzyme poly(ADP-ribose) polymerase-1 (PARP-1) and inhibition of respiration by NO are other likely mechanisms for NO-mediated energy failure and neurotoxicity (Moncada and Bolanos 2006).
NO activates Nrf2 through S-nitrosylation of cysteine residues residing on Keap1 in cultured rat pheochromocytoma cells. This chemical modification results in dissociation of Keap1 from Nrf2 and allows Nrf2 to translocate from the cytosol to the nucleus, where it binds to ARE and facilitates the transcription of antioxidant proteins. Accumulating evidence suggests that at low concentrations, NO plays a role in neurotransmission, vasodilation, and neuroplasticity, while at higher concentrations, NO has been implicated in the pathogenesis of stroke , demyelination, and other neurodegenerative diseases (Pacher et al. 2007).
In addition to proteins, NO also interacts with unsaturated fatty acids generating nitro-fatty acids, which are highly reactive electrophilic compounds that can modulate a variety of cellular targets (Jain et al. 2008; Trostchansky and Rubbo 2008). Examples of nitro-fatty acids include nitro-oleic acid, nitro-arachidonic acid, nitro-hydroxyeicosatrienoic acids (Fig. 6.6) (Trostchansky et al. 2007; Balazy et al. 2001; Gorczynski et al. 2009; Schopfer et al. 2002) Nitro-fatty acids have been shown to prevent inflammatory and atherogenic responses in endothelial cells (Hwang et al. 2009) as well as other cell types (Lim et al. 2002; Cui et al. 2006). Main effects of nitro-fatty acids include modulation of neutrophil and macrophage inflammatory responses (inhibition of cytokines, chemokines, and inducible enzymes involved in oxidative response) (Lim et al. 2002; Cui et al. 2006), inhibition of platelet aggregation (Coles et al. 2002), induction of antioxidant response (hemeoxygenase-1) (Wright et al. 2009; Iles et al. 2009), and increase in endothelium-independent vascular relaxation (Lima et al. 2005). Molecular mechanisms associated with protective actions of nitro-fatty acid are not fully understood. However, it is reported that nitro-fatty acids bind to nuclear receptors (i.e.,peroxisome proliferator-activated receptor-γ) and form adducts with proinflammatory transcription factors (i.e., NF-κB) leading in alterations in cell signaling processes (Li et al. 2008; Freeman et al. 2008; Schopfer et al. 2005).
6.7.2 Generation of Peroxynitrite and its Effect on Proteins, Lipids and Nucleic Acids
Under pathophysiological conditions NO reacts with superoxide anions to produce highly reactive peroxynitrite (ONOO−) . Like ROS, ONOO− produces oxidative damage proteins, lipids and nucleic acids (Fig. 6.7) (Beckman and Koppenol 1996; Koppenol 2001; Pacher et al. 2007; Radi 2009). At high concentrations peroxynitrite produces NO-mediated damage to biological molecules through oxidative and nitrosative stress (Pacher et al. 2007). Thus, the direct reaction of ONOO− with transition metal centers is among the fastest reaction that occurs in vivo. ONOO− modifies proteins containing a heme prosthetic group, such as hemoglobin, myoglobin, and cytochrome c, oxidizing ferrous heme into the corresponding ferric forms (Pacher et al. 2007). Similarly, peroxynitrite can inactivate inducible NOS by oxidative modification of its heme group (Huhmer et al. 1997), a reaction that may be responsible for negative feedback negative control of peroxynitrite generation under inflammatory conditions (Pacher et al. 2007). Peroxynitrite not only reacts rapidly with iron-sulfur clusters producing inactivation of mitochondrial aconitase and phoshogluconate dehydratase, but also inactivates Zn2 + sulfur motifs containing eNOS and alcohol dehydrogenase (Pacher et al. 2007) .
Related posts:
 Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Summary, Perspective and Direction for Future Studies
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Summary, Perspective and Direction for Future Studies
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


