Fig. 7.1
Long-term consumption of Western diet results in the increased production of ROS and neuroinflammation. Tricarboxylic cycle (TCA cycle), Platelet activating factor (PAF), Arachidonic acid (ARA), Nitric oxide (NO), Spinal cord injury (SCI), Traumatic brain injury (TBI), Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HT), and Amyotrophic lateral sclerosis (ALS)
Endothelial cells in cerebrovasculature also contribute to oxidative stress and damage in the brain. Endothelial cells, which perform a vital function in cerebrovascular system also predispose the brain to oxidative stress (Donato et al. 2007; Freeman and Keller 2012). For example, ROS-mediated oxidative damage within the vasculature promotes deleterious changes in blood-flow and blood brain barrier integrity, which initiate and induce remarkable neurochemical alterations (upregulation of nuclear factor-kappaB (NF-κB) and activator protein 1 (AP-1), increase in the gene expression of TNFα, IL-1β and IL-6 and adhesion molecules (ICAM-1 and VCAM-1) in neural cells leading to pathophysiology of neurotraumatic and neurodegenerative diseases (Farooqui 2010).
7.2 Effect of Hyperlipidic Diet on Non-Neural Tissues
Hyperlipidic diet is known to increase carnitine-palmitoyltransferase-1 (CPT-1) activity in the liver and muscle tissues (Ryu et al. 2005). The increase in muscle CPT-1 activity, due to high-fat diets rich in unsaturated fatty acids causes a reduction in CPT-1 sensitivity to malonyl-coenzyme A (malonyl-CoA). Synthesized from acetyl-CoA carboxylation, malonyl-CoA is critical for the regulation of lipid metabolism. It provides acetyl-activated groups to fatty acid chain synthesis and inhibits CPT-1 and, consequently, β-oxidation occurs. The high intake of unsaturated fatty acids augments mitochondrial membrane fluidity and reduces CPT-1 inhibition by malonyl-CoA (Liu et al. 2007). The fatty acid flux is increased in mitochondria interferes with β-oxidation and functioning of TCA cycle (Koves et al. 2008). Thus, fatty acids and their metabolites accumulate in mitochondria leading to mitochondrial stress and insulin resistance . In adipose tissues high levels of fatty acids in Western diet not only modulate gene expression through the involvement of nuclear receptors like peroxisome proliferator-activated receptors (PPARs), which act like transcription factors . It is reported that PPARs aid glucose uptake regulation via adiponectin, a protein expressed mainly by white adipose tissue. Their levels are reduced in various types of obesity and insulin resistance (Lebovitz and Banerji 2005). High-fat diet intake also exerts its effects upon gene expression of PPAR γ coactivator 1α. PPAR γ coactivator 1α consists of a PPAR coactivator, and both have a strict relationship with adipocyte synthesis and apoptosis along with increase in insulin sensitivity. In addition, fatty acids in Western diet modify messenger ribonucleic acid stability, but also alter de novo synthesis of the transcription factor, altering protein synthesis (Duplus et al. 2000; Ou et al. 2006).
7.3 Induction of Nitrosative Stress
Nitric oxide (NO) is another reactive specie that is generated by the mitochondria. In mitochondria, NO is generated during the breakdown of arginine to citrulline by a family of NADPH-dependent enzymes called mitochondrial nitric oxide synthases (mtNOS) . This isoform of NOS is responsive to changes in calcium concentration in the matrix and to play an important role in modulating mitochondrial respiration (Alvarez et al. 2003). Once formed, NO inhibits respiration by binding to heme groups in the proteins of the electron transport chain, including cytochrome c oxidase (Mason et al. 2006; Sarti et al. 2012). Thus, NO is a known inhibitor of the respiratory chain. NO competes directly with O2 at complex IV, reversibly retarding the formation of this complex and inducing ROS generation (Quintero et al. 2006). NO also inhibits complex I through S-nitrosylation (Burwell et al. 2006; Murphy 2009). This is more likely to occur in the settings of glutathione depletion reducing mitochondrial ROS production. Interactions between NO and complex IV occur rapidly and reversibly, playing an important role in short-term regulation of respiration and ROS production, whereas complex I inhibition by nitroso compounds is long lasting, indicating a greater role in long-term regulation of respiration and ROS generation. During high oxidative stress NO reacts rapidly with excess superoxide to form peroxynitrite, which can irreversibly inhibit multiple complexes of the respiratory chain, as well as dismutase enzymes, leading initially not only to elevation in oxidative stress and increase in Δψm (Geng et al.1992), but also causing alterations in calcium homeostasis, peroxidase enzymes (Murphy 2009).
It is well known that in the muscular, the adaptability of muscles to various external stimuli not only depends on efficient blood flow regulation, but also on integrity of vascular system. The blood flow to muscle cells is typically regulated by nitric oxide (NO), which is synthesized from arginine by nitric oxide synthase (NOS) . Homocysteine (Hcy) has been reported to reduce the bioavailability of NO through uncoupling of NOS and decreasing the uptake of arginine by the cells. Both of these mechanisms enhance ROS production. As stated above, in the presence of elevated ROS, NO reacts with ROS and generates peroxynitrite limiting NO signaling (Steed and Tyagi 2011). Hence it is conceivable that excess Hcy may compromise NO signaling and limit hemodynamics in muscular vessels leading to muscle fatigue, ischemia and reduction in physical endurance.
Consumption of Western diet results in not only dysregulation of ROS and RNS production in a variety of tissues, but is also linked with a number of inflammatory and age-associated neurological disorders with oxidative stress. These diseases include stroke , Alzheimer disease (AD) , and depression (Fig. 7.1) (Tidball and Wehling-Henricks 2007; Farooqui et al. 2012; Farooqui 2013). It is proposed that overproduction of O2 •− due to abnormal reduction of key components of the respiratory chain (i.e., ubiquinone and cytochrome b) or to impairment of antioxidant defenses adversely affects various cellular processes and constituents promoting oxidative stress in above mentioned chronic diseases (Rustin 2002; Droge 2002). In addition, reaction between O2 •− and NO generates peroxynitrite. This metabolite produces a variety of toxic effects in the brain.
7.4 Neurochemical Effects of Enzymic and Non-Enzymic Lipid Mediators of Arachidonic Acid Metabolism
It is well known that the brain is highly enriched in ARA, which is mostly esterified in the stereospecifically numbered-2 position of neural membrane phospholipids. ARA is essential for mediating neuroreceptor signaling . Stimulation of ARA signaling by glutamatergic, serotonergic, cholinergic or dopaminergic neuroreceptors triggers ARA release by AA-selective Ca2+-dependent cytosolic phospholipase A2 (cPLA2) . The released ARA is oxidized by cyclooxygenases (COXs) ; lipoxygenases (LOXs) , and epoxygenases (EPOXs) resulting in the formation of prostaglandins (PGs), leukotriene (LTs), lipoxins (LXs), and thromboxanes (TXs), as well as hydroxyl-eicosatetraenoic acid (HETE) and epoxyeicosatetraenoic acids (EETs), and dihydroxy-eicosatrienoic acids (DHETs) (Phillis et al. 2006; Farooqui and Horrocks 2007). Most ARA-derived lipid mediators produce prooxidant, prothrombotic, proaggregatory, and proinflammatory effects. However, lipoxin produces anti-inflammatory effects. Many eicosanoids contain unsaturated cyclopentenone ring structures. Due to the presence of reactive carbonyl moiety, cyclopentenone containing metabolites rapidly form Michael adducts with cellular thiols, including those found in glutathione (GSH) and proteins. These metabolites are collectively known as eicosanoids. Eicosanoids produce a wide range of biological actions including potent effects on neuroinflammation , vasodilation, vasoconstriction, apoptosis and immune responses (Phillis et al. 2006). At the molecular level, eicosanoids act within neurons to modulate the activities of ion channels, protein kinases, ion pumps, and neurotransmitter uptake systems . Many eicosanoids have been shown to exit the cell of their origin and act at a distance, by interacting with G-protein-coupled receptors (eicosanoid receptors) present on nearby neurons or glial cells. Finally, the actions of the eicosanoids may be terminated either by diffusion, uptake into phospholipids, or enzymic degradation (Farooqui 2011). Other eicosanoids, such as 15-deoxy-Delta(12,14)-PGJ2 (15d-PGJ2) and PGI2 act as a high affinity ligand for the nuclear receptor PPARγ and PPARδ to modulate gene transcription by binding to this receptor (Straus and Glass 2001). Other activities of the cyclopentenone PG are mediated by the reactive α,β-unsaturated carbonyl group located in the cyclopentenone ring. The transcription factor NF-κB and its activating kinase are key targets for the anti-inflammatory activity of 15d-PGJ2, which inhibits NF-κB-mediated transcriptional activation by PPARγ-dependent and independent molecular mechanisms (Straus and Glass 2001) .
Free radical species attack double bonds of free or esterfied ARA produces alkyl radicals, which in turn reacts with molecular oxygen to form a peroxyl radical (ROO•). Peroxyl radical can abstract hydrogen from adjacent PUFAs to produce a lipid hydroperoxide (ROOH) and a second alkyl radical, thereby propagating a chain reaction of lipid oxidation (Echtay 2007; Farooqui 2011). Breakdown of lipid peroxides results in the formation of α,β-unsaturated aldehydes including 4-hydroxynonenal (4HNE) , malondialdehyde (MDA) , and acrolein (Ac) (Fig. 7.2) (Esterbauer et al. 1991; Kehrer and Biswal 2000). These metabolites are diffusible and highly reactive with other biomolecules and, consequently, neurotoxic. The α,β-Unsaturated aldehydes covalently bind to proteins through reaction with thiol groups and alter their function. Their levels are markedly elevated in neurotraumatic and neurodegenerative diseases (Table 7.1). They also react with amino groups to form cyclic adducts. The α,β-unsaturated aldehyde interacts with proteins resulting in post-translational modifications. This process is called as protein carbonylation. The degradation of carbonylated proteins occurs via at least two different mechanisms. Some carbonylated proteins are degraded by proteasome independent mechanism, while other carbonylated proteins are metabolized in proteasome through the 26 S ubiquitination (Carbone et al. 2004) .
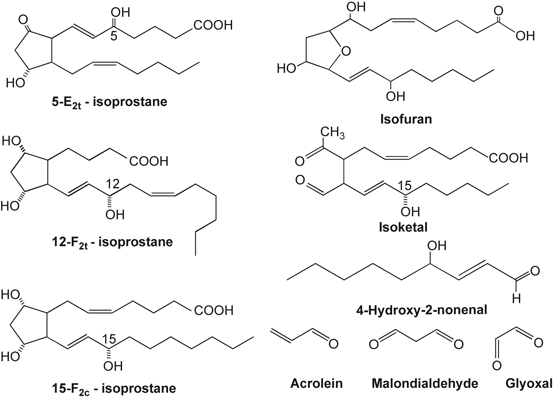
Fig. 7.2
Chemical structures of arachidonic acid-derived isoprostanes, isoketal, 4-hydroxynonenal, acrolein, malondialdehyde, and glyoxal
Table 7.1
Levels of α,β-unsaturated aldehydes (4-HNE, acrolein, and MDA) in neurological disorders
Neurological disorder | Levels of α, β-unsaturated aldehyde | Reference |
|---|---|---|
Stroke | Increased | Eaton et al. 1999 |
Traumatic brain injury | Increased | Zhang et al. 1999 |
Spinal cord injury | Increased | |
Alzheimer disease | Increased | |
Parkinson disease | Increased | Selley 1998 |
Amyotrophic lateral sclerosis | Increased | |
Multiple system atrophy | Increased | Shibata et al. 2010 |
Prion diseases | Increased | Andreoletti et al. 2002 |
Free radical species attack on lipid hydroperoxides also result in the synthesis of isoprostanes (IsoPs) via β-cleavage of the peroxyl acid and subsequent molecular rearrangement. IsoP contain D-, E-, and F-ringed structures similar to cyclooxygenase-generated prostaglandins, except that their hydrocarbon chains are in the cis position in relation to the pentane ring as opposed to the trans position observed in prostaglandins (Fam and Morrow 2003; Farias et al. 2008; Farooqui 2011). The estimation of isoP is used as a “gold standard” to quantify cumulative oxidative stress in neurotraumatic and neurodegenerative diseases. However, their neurochemical activities are poorly understood (Musiek et al. 2006; Zeiger et al. 2009; Farooqui 2011; Milne et al. 2013). Levels of IsoP are markedly increased in neurotraumatic and neurodegenerative diseases (Table 7.2). In addition, free radical attack also results in the formation of small amonts of isofurans (IsoF) and isoketals IsoK) (Fessel et al. 2003). Like IsoP, very little is known about neurochemical activities of IsoF and IsoK (Fig. 7.2). Among lipid peroxidation products, reactive α, β-unsaturated aldehydes and IsoPs have been reported to contribute to vascular disease and other oxidative stress-related pathologies not only by covalently modifying proteins and affecting critical protein functions, but also through receptor dependent mechanisms involving disruption of cell signaling and mitochondrial dysfunction (Farooqui 2011) .
Table 7.2
Levels of isoprostanes in neurotraumatic and neurodegenerative diseases
Neurological disorder | Levels of isoprostanes | Reference |
|---|---|---|
Stroke | Increased | Zeiger et al. 2009 |
Traumatic brain injury | Increased | Bayir et al. 2002 |
Spinal cord injury | Increased | Oner-Iyidoğan et al. 2004 |
Alzheimer disease | Increased | Montine et al. 2007 |
Huntington disease | Increased | Montine et al. 1999 |
Multiple sclerosis | Increased | Greco et al. 2000 |
Creutzfeldt-Jakob disease | Increased | Greco et al. 2000 |
Scrapie-infected mice | Increased | Minghetti et al. 2000 |
7.4.1 4-Hydroxynonenal and Its Contribution to Oxidative Stress-Mediated Injury
The oxidation and subsequent β-cleavage of ARA by hydroxyl radicals (OH•) yields 4-HNE, a nine carbon α, β-unsaturated aldehyde. 4-HNE is one of the major end products of lipid peroxidation and an important mediator of neural cell damage because of its ability to covalently modify biomolecules with disruption of important cellular functions (Esterbauer et al. 1991; Lin et al. 2005; Farooqui and Horrocks 2006) . The C3 position of 4-HNE is a highly reactive because of the presence of electeron withdrawing functional groups, the double bond. It serves as a site that undergoes a Michael addition reaction with cellular thiols cysteine, the imidizole nitrogen of histidine, and to a lesser extent, the amine nitrogen of lysine. The chemical reactions between 4-HNE and proteins include reactions between the C = C double bond with a nucleophile (Cys, glutathione (GSH) and amine) via 1,2- and 1,4-Michael addition. The 1,2-Michael addition involves the reaction of a primary amine (Lys) with the α,β-unsaturated carbonyl, resulting in the formation of a Schiff base at acidic pH (Petersen and Doorn 2004). 4-HNE not only inhibits DNA and RNA synthesis and disturbs calcium homeostasis, but also plays a substantial role in the disruption of the energy-producing capacity of mitochondria (Esterbauer et al. 1991; Guichardant et al. 2002). The localized concentration of 4-HNE may reach as high as 4.5 mM within the phospholipid bilayer. The half-life of 4-HNE is relatively short within cells, and 4-HNE rapidly removed by phase II reactions, allowing for excretion of water soluble conjugates (Gueraud et al. 2010) . Due to their amphiphilic nature, 4-HNE can easily diffuse across membranes and can covalently modify any protein in the cytoplasm and nucleus, far from their site of origin (Negre-Salvayre et al. 2008). Similarly, the 4-HNE formed outside the cells (i.e., in a site of inflammation or in plasma), can react with adjacent cells, even in cases when they are not primary sites of lipid peroxidation. 4-HNE modified proteins can also be removed by autophagic and proteasomal degradation pathways (Hill et al. 2008; Botzen and Grune 2007). The modification of adenine nucleotide translocator by 4-HNE causes the inhibition of enzymic activities and suppression of ADP and ATP transport through the inner mitochondrial membrane (Picklo et al. 1999). These events contribute to the disruption of the energy-producing capacity of mitochondria. In metabolic diseases, such as diabetes and metabolic syndrome, elevations in 4-HNE promote insulin resistance (Vincent et al. 2001; Russell et al. 2003). Moreover, at low doses, 4-HNE exerts an anti-cancer effect, by inhibiting cell proliferation, angiogenesis, cell adhesion and by inducing differentiation and/or apoptosis in various tumor cell lines (Pizzimenti et al. 2013). Collective evidence suggests that 4-HNE not only forms adducts with DNA (Esterbauer et al. 1991; Farooqui 2011; Chung et al. 2000), but also reacts with phospholipids (containing PUFAs such as linoleic and ARA) and nucleophilic amino acids (e.g cysteine, histidine and lysine residues (Farooqui 2011), making lipid membranes particularly vulnerable to 4-HNE modification .
The relative abundance of 4-HNE in the brain, vasculature, and adipose tissues depends not only on the rate of lipid peroxidation and 4-HNE synthesis, but also on the removal of 4-HNE adducts by phase II metabolic pathways such as glutathione-S-transferases. Thus, 4-HNE is metabolized by glutathione S-transferase A4 (GSTA4) producing glutathionyl-HNE (GS-HNE) and its metabolite glutathionyl-1,4-dihydroxynonene (GS-DHN). In 3T3-L1 adipocytes, high glucose levels and oxidative stress induce production of GS-HNE and GS-DHNE in a GSTA4-dependent manner and both glutathionylated metabolites induced secretion of TNFα from RAW264.7 and primary peritoneal macrophages. Targeted microarray analysis shows that GS-HNE and GS-DHN upregulates the expression of inflammatory genes (C3, C4b, c-Fos, igtb2, Nfkb1, and Nos2) supporting the view that GS-HNE and GS-DHN are pro-inflammatory glutathione metabolites of 4-HNE, which promote inflammation and insulin resistance in visceral tissues (Frohnert et al. 2013). Depending on its relative concentration, 4-HNE can induce a range of effects in the brain, vascular endothelial cells, adipose tissues, and smooth muscle cells. 4-HNE is also involved in carbonylation of adipocyte fatty acid-binding protein, a protein that has been implicated in the regulation of insulin resistance in vivo. In vitro, modification of adipocyte fatty acid-binding protein with 4-HNE has been mapped to Cys-117. Carbonylation of adipocyte fatty acid-binding protein results in10-fold reduction in fatty acid binding affinity for fatty acids. It is proposed that an increase in the carbonylation of a number of adipose-regulatory proteins may serve as a mechanistic link among increased oxidative stress, development of insulin resistance, and obesity (Grimsrud et al. 2007) .
Cross-linking of 4-HNE with glucose transporter, glutamate transporter, Na+, K+-ATPases, and NADP+-dependent isocitrate dehydrogenase produces changes in their activities (Mark et al. 1997; Keller and Mattson 1998; Lauderback et al. 2001; Yang et al. 2004). Inhibition of Na+, K+-ATPase by 4-HNE facilitates the opening of NMDA receptor channel and influx of calcium ions into the cell (Kadoya et al. 2003) (Fig. 7.3). This calcium entry can be very harmful for neurons. 4-HNE not only inhibits rat brain mitochondrial respiration, but also blocks neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin, which may contribute to the cytoskeletal changes in neurons undergoing a neurodegenerative process (Neely et al. 1999). In some cases, 4-HNE-mediated modification increases the activity of key regulatory proteins such as the dimerization and ligand-independent activation of the epidermal growth factor receptor (Liu et al. 1999) or the activation of the Nrf2 (nuclear factor erythroid 2-related factor 2) transcription factor, leading to increased expression of genes implicated in the antioxidant response (Zhang et al. 2006; West and Marnett 2005). In addition to altering the activity of enzymes, 4-HNE-mediated alkylation alters the rate of degradation of some proteins (alcohol dehydrogenase and αB-crystallin) (Carbone et al. 2004; Marques et al. 2004) (21, 22) .
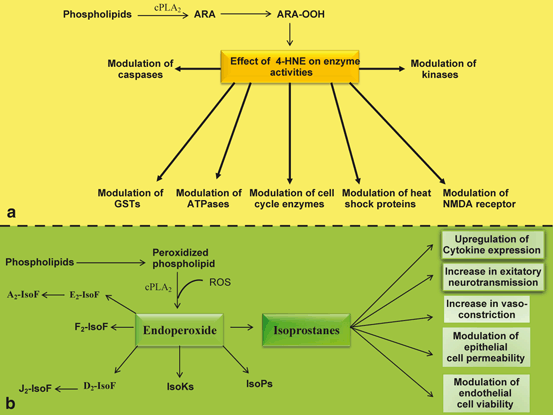
Fig. 7.3
Generation of 4-HNE and IsoP from arachidonic acid and neurochemical activities of 4-HNE and IsoP. a 4-HNE is generated from free arachidonic acid. Cytosolic phospholipase A2 (cPLA 2 ), 4-hydroxynonenal (4-HNE), and glutathione-S-transferase (GST). b Isoprostanes are derived from oxidized phospholipids. isoprostane (IsoP), and Isofurans (IsoF)
As stated above, 4-HNE interacts with reduced glutathione to form GS-HNE complex, which is reduced to GS-1,4-dihydroxynonanol (GSDHN). This complex along with protein kinase signaling cascade facilitates the migration of NK-κB from cytosol to the nucleus, where it binds to the NFkB DNA binding sequence and transcribes various genes for proinflammatory cytokines , chemokines , proinflammatory enzymes, and other inflammatory markers (Fig. 7.4). The expression of a large number of genes involved in apoptosis, cell growth, survival, differentiation, and immune response is regulated by NF-κB, which is associated with an array of diseases such as neurotraumatic, neurodegenerative, and autoimmune diseases. The migration of NF-κB is the key for 4-HNE to modulate neuroinflammation . 4-HNE is also involved in neurogenic inflammation and pain through activation of irritant transient receptor potential ankyrin1 (TRPA1) channel (Trevisani et al. 2007). 4-HNE-mediated inactivation of thioredoxin and thioredoxin reductase through modification of cysteine and selenocysteine residues at the active site has also been linked to dysregulation of cellular redox status (Fang and Holmgren 2006).
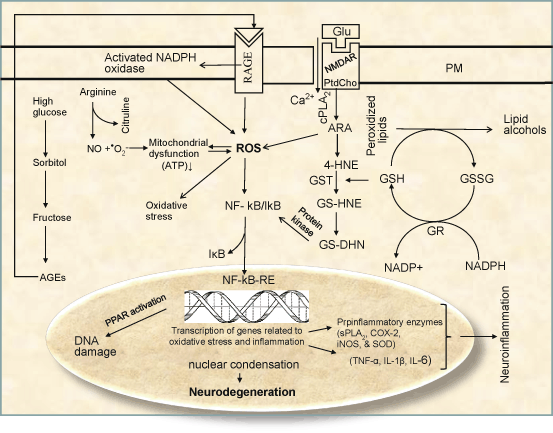
Fig. 7.4
Diagram showing the conjugation of GST with 4-HNE and activation of NF-κB by ROS. Glutamate (Glu), Plasma membrane (PM), Phosphatidylcholine (PtdCho), Arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), Platelet activating factor (PAF), N-methyl-D-aspartate receptors (NMDA-R), Cytosolic phospholipase A2 (cPLA 2 ), Cyclooxygenase (COX), Lipoxygenase (LOX), Reactive oxygen species (ROS), Nuclear factor-kappa B (NF-κB), Nuclear factor-kappa B response element (NF-κB-RE), Tumor necrosis factor-alpha (TNF-α), Interleukin-1beta (IL-1β), Interleukin-6 (IL-6), Nitric oxide (NO), Peroxynitrite (ONOO − ), Advanced glycation end-product (AGE), Receptors for advanced glycation end-product (RAGE), Reduced glutathione (GSH), Oxidized glutathione (GSSG), Glutathione reductase (GS), Glutathione-1,4-dihydroxynonene conjugate (GS-DHN), Glutathione-HNE conjugate (GS-HNE), Glutathione-S-transferase (GST)
Low levels of free 4-HNE (0.3 to 0.7 µM) have been reported to occur in the plasma of healthy individuals (Selley et al. 1989). 4-HNE levels increase significantly in plasma and tissues during aging (Gil et al. 2006) and in diseases associated with oxidative stress, such as atherosclerosis and diabetes, and neurological disorders (Farooqui 2011, 2013). Studies on the effect of 4-HNE on non-neural cells (vascular endothelial and smooth muscle cell) redox signaling and function have indicated that abundance of 4-HNE is found in the vasculature. Levels of 4-HNE in vasculature are not only dependent on the rate of lipid peroxidation and 4-HNE synthesis, but also on the removal of HNE adducts by phase II metabolic pathways such as glutathione-S-transferases (Farooqui 2011). Exposure of adipocytes to exogenous 4-HNE in a dose-dependent manner decreases in intracellular levels of adiponectin (Wang et al. 2012) . The expression of adiponectin gene expression is also elevated after 4-HNE treatment, with concomitant increase in peroxisome proliferator-activated receptor gamma (PPAR-γ) gene expression and transactivity. This effect can be abolished by T0070907, a PPAR-γ antagonist, supporting the view that PPAR-γ activation plays a critical role in this process (Wang et al. 2012). Based on studies on the effect of cycloheximide on adiponectin degradation, it is proposed that 4-HNE exposure enhances adiponectin protein degradation via ubiquitin-proteasome system. Collective evidence suggests that ARA-derived 4-HNE differentially regulates adiponectin gene expression and protein abundance and may play a mechanistic role in obesity-related plasma adiponectin decline (Wang et al. 2012). Exercise and dietary energy restrictions not only reduce 4-HNE levels, but also improve insulin sensitivity in muscle cells and increase levels of 4-HNE detoxifying systems (glutathione and oxidoreductases). Depending on its relative concentration, 4-HNE produces a range of hormetic effects in vascular endothelial and smooth muscle cells, including kinase activation, proliferation, induction of phase II enzymes and in high doses inactivation of enzymic processes and apoptosis (Farooqui 2011). 4-HNE also plays an important role in the pathogenesis of vascular diseases such as atherosclerosis, diabetes, and neurodegenerative diseases (Farooqui 2011) .
7.4.2 Isoprostanes and Their Contribution to Oxidative Stress-Mediated Injury
As stated above, IsoP are prostaglandin-like compounds generated by non-enzymic oxidation of esterified polyunsaturated fatty acids (arachidonic, eicosapentaenoic, adrenic acids) in neural membrane phospholipids. They are released by the action of cPLA2 on peroxidized glycerophospholipids (Fig. 7.3) . The molecular mechanism by which isoprostanes are formed is analogous to the synthesis of prostaglandins by cyclooxygenases (Morrow et al. 1999). Unlike prostaglandins, which are formed from the COX, LOX, and EPOX-mediated oxidation of free ARA, the formation of isoprostanes in situ is initiated at the esterified ARA on the glycerophospholipid molecule (Fam and Morrow 2003). IsoPs are formed in situ in the phospholipid domain of cell membranes and circulating lipoproteins. They are then cleaved by phospholipases A2, released extracellularly, circulate in blood and are excreted in urine.
Non-enzymic synthesis of the family of F2-isoprostanes involves the formation of positional peroxyl radical isomers of ARA, which undergo 5-exo cyclization and a second molecule of oxygen adds to the backbone of the compound to form PGG2-like compounds. These unstable bicycloendoperoxide intermediates are then reduced to the F2-IsoPs. F2-Isoprostane (F2-IsoP) that is subsequently released in free form by the action of PLA2 (Morrow et al. 1991; Fam and Morrow 2003; Montuschi et al. 2007) (Fig. 7.3). Another mechanism of isoprostane generation starts with a 4-exocyclization of a peroxyl radical leading to an intermediate dioxetane (Durand et al. 2005). Formation of F2-IsoPs, E2– and D2-IsoPs can be obtained by rearrangement of H2-IsoP endoperoxides. The occurrence of E2/D2-IsoPs has not been reported during ischemic injury (Farias et al. 2008). One of the E2-IsoP isomers, 8-iso–PGE2, which is produced abundantly in vivo, has a very potent biological activity and thus may contribute to brain damage associated with oxidative stress. As stated earlier, measurement of isoP is considered to be one of the most reliable index for assessing lipid peridation in vivo .
Although, IsoPs are initially formed in vivo esterified in phospholipids, but most studies exploring their bioactivity have been performed using unesterified IsoPs. 15-A2t-IsoP is known to produce neurodegeneration in cultured neurons via mitochondrial ROS production, glutathione depletion, 12-lipoxygenase activation, and caspase cleavage (Musiek et al. 2006). Furthermore, 15-A2t-IsoP also potentiates hypoxia-induced neuronal cell death, supporting the view that IsoP may be closely associated with the pathogenesis of ischemic injury (Zeiger et al. 2009). In brain microvasculature isoPs act as vasoconstrictors by inducing COX-mediated synthesis of thromboxane in endothelial cells (Lahaie et al. 1998). They induce their biochemical effects both via receptor-dependent and independent mechanisms. IsoP binds with TxA2 receptors (TPRs) (Fig. 7.5). These receptors are not only found in the brain, but also in macrophages or monocytes, vascular endothelial cells, and platelets. TPRs modulate antiatherosclerotic, antivasoconstrictive, and antithrombotic effects, depending on the cellular target. TPR receptors mobilize intracellular Ca2+ and are coupled with the activation of protein kinases (MAP kinase and Ca2+/Rho kinase) (Kinsella et al. 1997; Pratico et al. 1997) . F2-IsoP and its receptor also mediate their effects in vascular beds and platelet function by promoting interactions between endothelial cells and monocytes (Lahaie et al. 1998; Fam and Morrow 2003). Isoprostane-mediated monocyte adhesion does not dependent on VCAM-1, but involves protein kinases such as protein kinase A and mitogen-activated protein kinase kinase 1. F2-IsoP also modulates the p38 MAPK pathway during monocyte adhesion (Cracowski 2004). Thus, F2-IsoP not only affects vascular and bronchial smooth muscles function, but also modulates cellular proliferation (Fam and Morrow 2003). These processes may relate to inflammation and atherosclerosis. Receptor independent action of F2-IsoP is due to adduct formation. These compounds contain reactive α,β-unsaturated carbonyl group on the prostane ring, which readily reacts with thiol-containing compounds to produce many biological effects. In isolated bovine retinae isoP produce dual effects. Low concentrations of 8-isoPGF2α inhibit; whereas, higher concentrations of 8-isoPGF2α stimulate K+-mediated [3H]D-aspartate overflow (Opere et al. 2005). Based on detailed investigations, it is suggested that both inhibitory and stimulatory effects are mediated via the activation of thromboxane receptors .
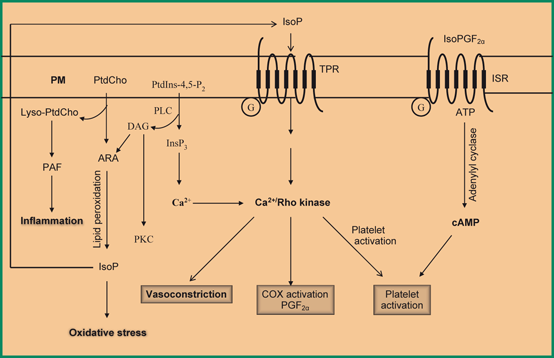
Fig. 7.5
Generation of isoprostane from arachidonic acid and stimulation of thromboxane receptor by isoprostane and isoPGF2α. TXA2receptor (TRP), Plasma membrane (PM), Phosphatidylcholine (PtdCho), Arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), Rho-associated kinase (Rho-kinase), and Cyclic AMP (cAMP)
4-HNE-mediated protein carbonylation and treatment with cyclopentenone prostaglandin, 15-deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) are known to upregulate transcriptional activation of antioxidant-response genes . Thus, Nrf2, a central transcription factor involved in the regulation of antioxidant-responsive element-containing genes is activated in response to oxidative stress caused by the generation of 4-HNE and 15d-PGJ2. The alkylation of the cytoplasmic inhibitor of Nrf2, Keap1 (Kelch-like ECH-associated protein 1), by 4-HNE and exposure of Nrf2-Keap1 complex to 15d-PGJ2 mediate the dissociation of the Keap1-Nrf2 complex (Fig. 7.6). Once freed from inhibition by Keap1, Nrf2 migrates to the nucleus, where it interacts with antioxidant response elements (ARE) and facilitates the transcription of heme oxygenase-1 (HO-1), glutathione synthase (GS), NADP(H) quinine-oxidoreductase1 (NQO1), catalase and Na+-independent cystine/glutamate exchanger. This process results in strengthening of antioxidant defenses of neural cells (Fig. 7.6) (Levonen et al. 2004). In addition, 15d-PGJ2 binds with peroxisome proliferator-activated receptor-gamma (PPAR-γ) and activates it in a dose-dependent manner by down-regulating NFκB (Ou et al. 2006). Both pathways decrease neuroinflammation (Giri et al. 2004). Post-infarct PGJ2 administration produces neuroprotective effects following middle cerebral artery occlusion (MCAO) by non-overlapping NFκB and PPAR-γ dependent and independent mechanisms (Pereira et al. 2006; Lin et al. 2006) as well as being anti-inflammatory when used pre-induction in models of mixed stroke and in non-neuronal systems. Collective evidence suggests that 15d-PGJ2 is effective in protecting against white matter ischemic in the brain .
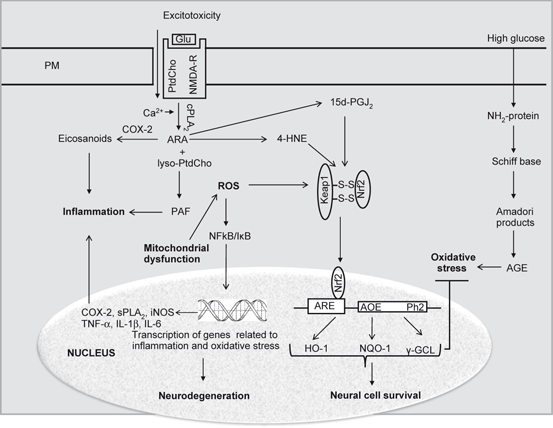
Fig. 7.6
Effect of ROS, 4-HNE and 15d-PGJ2 on Nrf2/Keap complex and migration of Nrf2 into the nucleus. Plasma membrane (PM), Phosphatidylcholine (PtdCho), Arachidonic acid (ARA), lysophosphatidylcholine (lyso-PtdCho), Platelet activating factor (PAF), N-methyl-D-aspartate receptors (NMDA-R), Cytosolic phospholipase A2 (cPLA 2 ), Cyclooxygenase (COX), Reactive oxygen species (ROS), Nuclear factor-kappa B (NF-κB), Nuclear factor-kappa B response element (NF-κB-RE), Tumor necrosis factor-alpha (TNF-α), Interleukin-1beta (IL-1β), Interleukin-6 (IL-6), Nuclear factor-erythroid 2-related factor 2 (Nrf2), Kelch-like ECH-associated protein 1 (Keap1), Hemeoxygenase (HO-1), NAD(P)H:quinone oxidoreductase-1 (NQO1), Antioxidant response element (ARE), γ-glutamate-cysteine ligase (γ-GCL), and Glutathione S-transferase (GST)
7.4.3 Isoketals and Their Contribution to Oxidative Stress-Mediated Injury
Recently, another class of chemically reactive lipid peroxidation products has been identified: γ-ketoaldehyde isoketals (IsoKs) derived from ARA This γ-ketoaldehydes is much more reactive with cellular nucleophiles than 4-HNE and, unlike the structurally similar COX-derived levuglandins, IsoKs remain esterified to phospholipids. Thus, the synthesis of isoketals occurs through the rearrangement of H2-IsoP endoperoxides. Isoketals differ from isoprostanes in containing a characteristic aldehyde group in a 1,4-dicarbonyl array, making them extremely reactive toward primary amino groups in proteins (Boutaud et al. 2005). Unlike F2-IsoP, isoketals result in modification of biologically important proteins rather than activation of specific receptors (Davies et al. 2004). Isoketals are highly reactive γ-ketoaldehydes that form pyrrole adducts with the ε-amino group of lysine residues on protein (Davies et al. 2004). These pyrrole adducts are unstable in the presence of oxygen and are further transformed to lactam and hydroxylactam adducts, which accumulate as stable end products. IsoKs adduct almost instantaneously to protein lysine residues and readily induce protein-protein cross-linking. IsoK-mediated protein cross-linking induces impairment in proteasomal degradation of adducted proteins and inhibiting proteasome function. These processes may result in disruption of blood brain barrier and neuronal dysfunction, which may ultimately lead to dementia (Davies et al. 2004). In addition, isoketals have remarkable ability to crosslink proteins through oxidation of the pyrrole. Isoketals have been detected in tissues as well as biological fluids. In non-neural cells, IsoK also reacts with phosphatidylethanolamine (PTdEtn) and DNA (Sullivan et al. 2010; Carrier et al. 2009) . Exogenous addition of IsoK to human umbilical vein endothelial cells (HUVEC) produces approximately five-fold more abundant IsoK-PE adduct than protein adduct (Sullivan et al. 2010) Nothing is known about the levels and roles of IsoK-PE in vivo.
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Neurochemical Aspects of Oxidative and Nitrosative Stress
 Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
 Effect of Exercise on Oxidative Stress in Neurological Disorders
Effect of Exercise on Oxidative Stress in Neurological Disorders
 Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
 Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
 Contribution of Dietary Fat in Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


