Fig. 5.1
Effects of exercise on various parameters related to changes in the brain
Since there is very little difference between physical activity (any activity that increases heart rate and energy expenditure from one’s basal level) and exercise (any structured or repetitive physical activity that is aimed to improve cardiovascular-respiratory fitness, muscle power and endurance, flexibility, agility, balance and/or body composition); therefore I have used the term exercise throughout this book. It is well known that exercise delays the onset of chronic diseases, such as diabetes, metabolic syndrome, cardiovascular and cerebrovascular diseases, neurodegenerative diseases, osteoporosis, and some cancers (Farooqui 2013). Molecular mechanisms associated with beneficial effects of exercise on chronic diseases are not fully understood. However, it is proposed that two mechanisms may contribute to the beneficial effects of exercise. One mechanism is associated with ROS sensitive pathway , which involves brain-derived neurotrophic factor (BDNF) , insulin-like growth factor-1 (IGF-1) , heat shock proteins (Hsp) (Allen and Tresini 2000), and peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α (Handschin and Spiegelman 2008) and the other mechanism involves the activation of redox-sensitive pathway resulting in gene products that are known to restore intracellular oxidant-antioxidant hemeostasis and protection from potential oxidative stress (Ji et al. 2009, 2010). These products include (a) antioxidant enzymes (e.g. manganese superoxide dismutase [MnSOD], glutathione peroxidase [GPX], and γ-glutamyl cysteine synthetase); (b) transcription factors and co-factors c-fos, activating transcription factor [ATF]-2, and PGC-1α; (c) redox status controlling molecules (e.g. thioredoxin, glutathione); and (d) proteins modulating metabolic status, such as uncoupling proteins (UCP), enzymes in fatty acid and glucose metabolism, and mitochondrial fusion and fission proteins (Ji et al. 2009, 2010). In addition, exercise also alters the molecular composition and architecture of cell membranes and extracellular matrix, which in turn plays an important role in conveying extracellular signals from the cell surface to the nucleus to induce a neurochemical response at the gene level. Alterations in neural membrane composition and extracellular matrix result in changes in neural membrane permeability and fluidity, which are closely associated with cell death, adaptation and survival. Accumulating evidence suggests that exercise promotes the prevention of chronic diseases and premature death (Lee and Sherrett 2001). However, controversies remain over the optimal “volume” (frequency, duration and intensity of exercise) and the minimum volume for health benefits of exercise, in particular the effects of intensity (e.g., moderate versus vigorous) on health status. There is evidence that intensity of physical activity is inversely and linearly associated with mortality (Lee and Sherrett 2001). Earlier studies have indicated that regular physical activity (expending > 2,000 kcal [8,400 kJ] per week) increases life expectancy by 1–2 years by the age of 80 and that the benefits are linear even at lower levels of energy expenditure. However, in recent years, most health and fitness organizations and professionals advocate a minimum volume of exercise that expends 1,000 kcal (4,200 kJ) per week and acknowledge the added benefits of higher energy expenditures (Paffenbarger et al. 1993; Lee and Sherrett 2001). It must be mentioned here that prolonged or high-intensity exercise produces harmful effects not only due to oxidative damage to macromolecules in both blood and skeletal muscle, but also through the activation of the hypothalamic-pituitary-adrenal (HPA) axis and the release of corticosterone from the adrenal cortex. Increased levels of corticosterone produce harmful effects (Shephard and Sidney 1975; Powers and Jackson 2008) .
5.2 Molecular Mechanisms Associated with the Effect of Exercise on the Brain
Regular exercise ameliorates age-related neuronal loss and produces positive effect on neurodegenerative diseases (Trejo et al. 2002). One of the regions of brain more affected by exercise is the hippocampus. Exercise increases the size of the anterior hippocampus, leading to improvements in spatial memory (Erickson et al. 2011). Exercise also increases the hippocampal volume by 2 %, effectively reversing age-related loss in volume (Persson et al. 2003a,b; Erickson et al. 2011). At the molecular level exercise up-regulates the expression of genes associated with the actions of BDNF , IGF systems, serotonin, and β-endorphins (Table 5.1). These factors not only regulate synaptic plasticity, and cell proliferation (Molteni et al. 2002a, b), but also control motivation, cognition, mood, and pain perception. Increase in hippocampal volume is associated with higher serum levels of BDNF and IGF, and other neurotrophic factors, which promote neurogenesis in the dentate gyrus (Erickson et al. 2011). The molecular mechanisms associated with increased expression of BDNF and IGF are not fully understood. However, it is proposed that exercise-mediated mild stress may stimulate influx of calcium ions, which may activate transcription factors that initiate the expression of the BDNF and IGF genes, creating BDNF and IGF proteins that may be involved in neurogenesis (Mattson et al. 2004) (Fig. 5.2). BDNF mediates its effects through interactions with two types of receptors, namely, the tyrosine kinase receptor (Trk) B receptor and the pan-neurotropin receptor p75 (p75NTR) (Reichardt 2006). The underlying molecular mechanisms driving the elevation of BDNF remain unknown. However, recent studies have indicated that exercise increases the synthesis of FNDC5, a membrane protein, which is highly expressed in heart and brain and less in skeletal muscle (Teufel et al. 2002). FNDC5 contains a signal peptide, fibronectin type III repeats, and a hydrophobic region, which encodes for a transmembrane domain. FNDC5 is cleaved and secreted as irisin, a hormone in the hippocampus of mice (Teufel et al. 2002; Wrann et al. 2013). Neuronal Fndc5 gene expression is regulated by PGC-1α, and Pgc1a-/- mice show reduction in Fndc5 expression in the brain. Furthermore, forced expression of FNDC5 in primary cortical neurons increases BDNF expression, whereas RNAi-mediated knockdown of FNDC5 reduces BDNF . Importantly, peripheral delivery of FNDC5 to the liver via adenoviral vectors not only produces elevation in blood irisin, but also induces expression of BDNF and other neuroprotective genes in the hippocampus (Wrann et al. 2013). Collectively, these studies indicate that exercise mediate its effect by stimulating the production of BDNF, which in turn stimulates the growth of new nerves and synapses and also preserves the survival of existing neurons. BDNF protein is synthesized as a precursor, pre-proBDNF protein, resulting after cleavage in a 32-kDa proBDNF protein. ProBDNF is either proteolytically cleaved intracellularly by enzymes like furin or pro-convertases and secreted as the 14 kDa mature BDNF (mBDNF), or secreted as proBDNF and then cleaved by extracellular proteases, such as metalloproteinases and plasmin, to mBDNF (Lessmann et al. 2003). Pro-BDNF preferentially interacts with the p75NTR, whereas mBDNF) selectively binds and activates the TrkB (Chao and Bothwell 2002; Ibanez 2002) and promotes long term potentiation. Interactions of mBDNF with protein tyrosine kinase receptor (TrkB) regulates a number of processes including memory formation, learning and behavior, synaptic plasticity (Soppet et al. 1991; Monteggia et al. 2004) (Fig. 5.3) through the involvement of various signaling pathways including the Ras/MAP and PtdIns 3K pathways. In contrast, binding of endogenous pro-BDNF with p75NTR is associated with long term depression in the hippocampus (Rosch et al. 2005; Wu 2005), and is associated with apoptotic cell death in peripheral neurons through the involvement of JNK (Fig. 5.3) (Teng et al. 2005). Accumulating evidence suggests that BDNF has emerged as the main chemical translator of the neuro-physiological effects of exercise on the intact brain (Vaynman et al. 2003). BDNF exerts its receptor mediated effects throughout the brain, including the support of neuronal survival, differentiation, and connectivity; it also plays a role in activity-dependent synaptic plasticity and is implicated in processes of learning and memory in normal individuals and in patients with neurological disease (Zhang and Ko 2009). The production/release of BDNF is strongly linked to the serotonin system and plays an essential role in mood and memory processes (van Donkelaar et al. 2009; Pollak et al. 2008). Moreover, brain tryptophan levels and serotonin metabolism correlate positively with BDNF in both prefrontal cortex and hippocampus, again highlighting the close interconnection and commonality between the underlying signaling systems (Esch and Stefano 2010) .
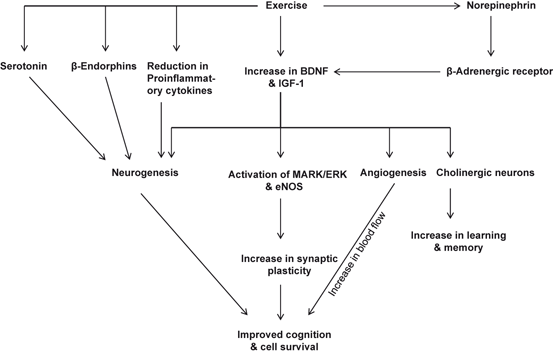
Fig. 5.2
Effect of exercise on the neurogenesis, angiogenesis, and synaptic plasticity
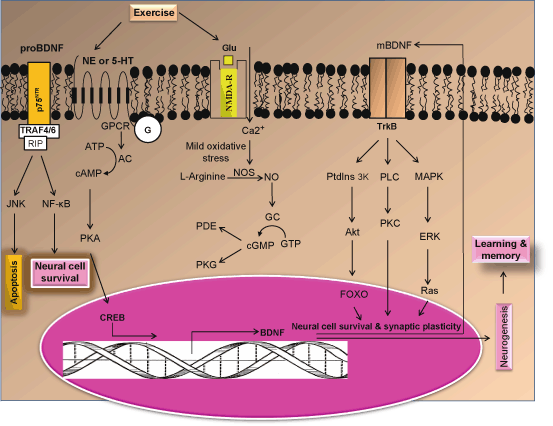
Fig. 5.3
Hypothetical diagram showing the contribution of norepinephrine or 5-hydroxytryptamine receptors in neuronal survival, neurogenesis, and learning and memory formation. NE Norepinephrine, 5-HT 5-hydroxytryptamine, PKA protein kinase A, CREB cAMP response element-binding protein, NOS nitric oxide synthase, NO nitric oxide, GC guanylate cyclase, AC adenylate cyclase, cGMP cyclic guanosine monophosphate, cAMP cyclic adenosine monophosphate, TrkB Tropomyosin receptor kinase B, PDE phosphodiesterase, PtdIns 3K Phosphatidylinositide 3-kinases, Akt a cytosolic protein kinase, MAPK Mitogen-activated protein kinases, ERK Extracellular signal-regulated kinases, GPCR G protein coupled receptor, NMDA-R N-methyl-D-aspartate receptors, BDNF brain-derived neurotrophic factor
Table 5.1
Exercise-mediated release of neurotransmitters, growth factors, and neuropeptide
Substances release from the brain | Reference |
|---|---|
Brain-derived neurotrophic factor | |
Insulin like growth factor-1 (IGF-1) | |
β-Endorphins | Persson et al. 2003a |
Serotonin | Klempin et al. 2013 |
Synapsin I | Vaynman et al. 2003 |
Synaptophysin | Vaynman et al. 2003 |
CAMK II | |
MAPK II |
IGF-1 is produced both in the brain and in the periphery in response to aerobic (Carro et al. 2001; Ding et al. 2006a) and resistance (Cassilhas et al. 2007) exercise. Furthermore, blocking entry of peripheral IGF-1 into the brain during aerobic training also prevents exercise-mediated hippocampal neurogenesis (Klempin et al. 2013), angiogenesis (Lopez-Lopez et al. 2004), and exercise-mediated brain injury recovery (Carro et al. 2001). It is also shown that blocking IGF-1 receptors in the hippocampus during exercise abolishes exercise-mediated increase in hippocampal BDNF mRNA and protein expression (Ding et al. 2006a, b). IGF-1 also modulates signal transduction process though a phosphatidyl-inositol-3 kinase/protein kinase B signaling targets rapamycin (PtdIns 3K/AKT/mTORc1) or mTOR pathway (Rommel et al. 2001). mTOR controls cell growth through two mechanisms: (a) one mechanism involves the mTORC1-mediated phosphorylation of ribosomal protein S6 kinase (S6K1) and 4E binding protein 1 (4EBP1) and the other mechanism is associated with mTORC1-induced increase in translational capacity through regulation of ribosomal RNA (rRNA) production within the nucleolus (Iadevaia et al. 2012).
Exercise-mediated increase in levels of vascular endothelial growth factor (VEGF) also induces neurogenesis (Fabel et al. 2003). The role of VEGF in neurogenesis has been confirmed in a set of experiments showing that blocking peripheral VEGF significantly inhibits exercise-induced neurogenesis (Fabel et al. 2003). In addition, by facilitating neovascularization VEGF also contributes to angiogenesis. Collective evidence suggests that increase in levels of BDNF, IGF-1, and VEGF play important roles in mediating the effects of exercise on brain health and performance through neurons and neurotransmitter .
Exercise also increases the release of β-endorphins into the circulation (Table 5.2). β-endorphin acts preferentially on μ opioid receptors (MOR), which are distributed throughout the brain (Ableiner and Schulz 1992; Colt et al. 1981; Hoffmann et al. 1990). Importantly, in vitro studies have shown that treatment of hippocampal progenitor cells with β-endorphin results in the activation of MOR (Persson et al. 2003a). Blockade of MOR by opioid antagonists causes decrease in MOR responses (Persson et al. 2003b) supporting the view that β-endorphins plays an important role in cell proliferation in vivo and they may be involved in the promotion of hippocampal neurogenesis through exercise (Fig. 5.2). β-Endorphins also modulate mood and pain perception. It is also reported that N2KO mice, which have a targeted deletion of the basic helix-loop-helix transcription factor, Nescient Helix-Loop-Helix 2 (Nhlh2) lack gene for Nhlh2. N2KO mice do not show an increase in β-endorphin secretion during or after exercise. In addition, N2KO mice also display adult-onset obesity with significant increases in their fat depots, abnormal responses to cold exposure, and reduced spontaneous physical activity levels (Good et al. 2008). Unlike other mouse obesity models, N2KO mice gain weight due to reduction in spontaneous physical activity, which precedes body weight gain and mild hyperphagia in older animals (Coyle et al. 2002) .
Table 5.2
Exercise induces the release growth factors, β-endorphins, serotonin, synaptic proteins, and enzymes from the brain
Substances release from the brain | Effect | Reference |
|---|---|---|
Brain-derived neurotrophic factor | Increased | |
Insulin like growth factor-1 (IGF-1) | Increased | |
β-Endorphins | Increased | Persson et al. 2003a |
Serotonin | Increased | Klempin et al. 2013 |
Synapsin I | Increased | Vaynman et al. 2003 |
Synaptophysin | Increased | Vaynman et al. 2003 |
CAMK II | Increased | |
MAPK II | Increased | |
Glutamine synthase | Increased | Bernardi et al. 2013 |
Glial fibrillary protein | Decreased | Bernardi et al. 2013 |
Exercise effectively promotes neurogenesis by effectively facilitating the release of serotonin (5-hydroxytryptamine) in the brain (Fig. 5.2). This neurotransmitter not only elevates mood during and after the workout, but also regulates, appetite, temperature and sleep/wake cycle (Fernstrom and Fernstrom 2006). Studies on tryptophan hydroxylase (TPH) 2 deficient (Tph2-deficient) mice demonstrate that lack of brain serotonin retards exercise-induced neurogenesis. Tph2-deficient mice exhibit normal baseline hippocampal neurogenesis but display impairment in exercise-induced neural cell proliferation. These studies suggest that exercise-mediated release of serotonin is required for adult hippocampal neurogenesis (Klempin et al. 2013). In addition, it is also reported that exercise also increases levels of norepinephrine (NE), which activates beta-adrenergic receptors, and may be closely involved in exercise-induced BDNF up-regulation (Fig. 5.2). The possible intracellular signaling pathways of NE-mediated BDNF up-expression may involve GPCR-MAPK-PtdIns-3K crosstalk and positive feedback (Ma 2009). Thus, expression and generation of BDNF, IGF, serotonin, NE, and β-endorphin are neuroprotective responses to exercise-mediated mild stress that not only generate new neurons, but also promote synaptic plasticity (the efficiency of signal transmission across the synaptic cleft between neurons, generally considered as the basis of learning and memory (Mattson et al. 2004). The combination of healthy diet and exercise produces additive effects on synaptic plasticity and cognitive function. The synthesis of ATP in mitochondria also promotes the activation of BDNF and IGF, which supports synaptic plasticity and cognitive function. Energy-balancing molecules (ubiquitous mitochondrial creatine kinase, AMP-activated protein kinase and uncoupling protein 2) interact with BDNF to modulate synaptic plasticity and cognition (Kramer et al. 1999). As mentioned above, healthy diet and exercise initiate and maintain energy metabolism in the visceral organs and brain. The autonomic nervous system sends information to the brain about various aspects of digestion such as motility, secretion, and blood flow. Vagal afferents are both sensitive to chemicals released in to blood as well as visceral distension and pain. Brain has the ability to integrate signals from visceral organs to modulate energy metabolism (i.e., feeding behaviors, food breakdown, energy acquisition, expenditure, utilization, storage, and transformation) (Gomez-Pinilla 2008) .
In addition, exercise also up-regulates genes related to the N-methyl-D-aspartate receptor (NMDA-R) function and down-regulates genes associated with the GABAergic system. In addition, genes for the expression of proteins associated with synapsin I and II, synaptotagmin, and syntaxin, calcium/calmodulin protein kinase II (CaM-KII), mitogen-activated protein kinase (MAPK/ERK, I and II), protein kinase C (PKC-δ); or transcription factor cAMP response element binding protein (CREB) are also increased (Vaynman et al. 2003; Molteni et al. 2002a, b; Ding et al. 2006a, b; Gomez-Pinilla 2008; Gomez-Pinilla et al. 2008). Exercise also stimulates the expression of several key intermediates of the PtdIns 3K/Akt pathway, which contribute to neuronal survival through the phosphorylation of a number of target proteins, such as GSK-3β, endothelial nitric oxide synthase (eNOS), the proapoptotic Bcl-2 family member BAD, caspase 9, the ubiquitin ligase murine double minute 2 (mdm2), and others. Phosphorylation of BAD targets it to 14-3-3 protein where it is sequestered, thereby blocking its proapoptotic role (Fayard et al. 2005; Chen and Russo-Neustdt 2007; Fleming et al. 2005). Thus, exercise mediates its effects through the activation of BDNF and IGF-mediated intracellular signal pathways involving protein kinases (MAPK/ERK and CaM-K), GSK-3β, and endothelial nitric oxide synthase (eNOS) (Vaynman et al. 2003; Vaynman et al. 2004; Molteni et al. 2002a, b). Additionally, cAMP/PKA-mediated activation of synapsin I phosphorylation facilitates regenerative growth of neurons and promotes neuronal survival. Exercise also upregulates the expression of the mitochondrial uncoupling protein 2, an energy-balancing factor involved in ATP formation and management of ROS (Vaynman et al. 2004, 2006). These observations support the view that in brain physical exercise-mediated release of BDNF not only regulates hippocampal synaptic plasticity, but also controls body weight and energy status by reducing body weight, normalizing glucose levels, and increasing insulin sensitivity (Gomez-Pinilla 2011). Blocking BDNF and IGF signaling abolishes the effects of exercise on neuroplasticity and learning and memory (Vaynman et al. 2004) .
Neuroprotective effects of calorie restriction are analogous to the beneficial effects of exercise on muscle and heart cells, where energy demand, ionic stress, and adaptive stress responses are increased during the exercise. Overconsumption of Western diet or strenuous exercise produces harmful effects in the muscles through the production of high levels of ROS and increased expression of cytokines and chemokines. In brain high levels of ROS suppress synaptic plasticity and decrease cognitive function by down-regulating the production of BDNF. In contrast, calorie restriction produces beneficial effects in the brain by modulating silent information regulator 1 (SIRT1) . SIRT1 not only reduces ROS, but also promotes chromatin modifications. These processes have positive effect on cognition (Laurin et al. 2001). Based on detailed investigations it is suggested that exercise and calorie restriction are metabolic stressors, which display a property known as “hormesis” , a process, which is defined as an adopted response to explain how a mild oxidative and nitrosative stress (production of ROS and RNS) associated with moderate exercise and calorie restriction can result in favorable adaptations that protect the body against more severe stresses and disorders derived from physical stress or other etiological origin (Ji et al. 2009; Nunn et al. 2010). Hormesis-mediated adaptations is supported not only by low levels of ROS and RNS production, but also by expression of heat shock protein responses, cell cycle regulation and apoptosis, DNA repair, fatty acid deacylation-reacylation, unfolded protein responses and autophagy stimulation (Farooqui et al. 2000).
Collectively these studies suggest that exercise and calorie restriction produce their beneficial effects not only by inducing neurogenesis, increasing hippocampal neural plasticity, and elevating cerebral blood flow, but also by maintaining the physiological redox state and normal functional connectivity of the brain. These processes are closely associated with healthy brain aging .
5.3 Molecular Mechanisms Associated with the Effect of Exercise on Neuroinflammation
Exercise improves overall health by inhibiting neuroinflammation and delaying the onset of common age-related chronic neural and non-neural diseases, including stroke , Alzheimer disease (AD) , Parkinson disease (PD) , diabetes, metabolic syndrome, and arthritis (Kujala 2009; Pedersen 2011) . As stated in chap. 1 and 2, the term ‘neuroinflammation’ refers to a multicellular process, which is characterized by (a) changes in local vasculature (increased blood flow and vascular permeability), (b) activation of resident microglial cells and astrocytes, (c) infiltration of mobile cells of the immune system (neutrophils, macrophages, and lymphocytes), and (4) increase in production of cytokines along with generation of a variety of lipid mediators, such as PGE2 and PAF (Graeber et al. 2011). Neuroinflammation is supported by several receptors (classical neurotransmitter and purinergic receptors) on neural cell surface found in microglial cell surface (Fig. 5.4). Microglial cells have been shown to express receptors for proteases, such as thrombin, which may come into play when an injury causes inundation of the parenchyma by plasma content but also when such factors are operating within the intact CNS (Balcaitis et al. 2003; Hanisch et al. 2004) . The activation of microglia induces both neuroprotective and neurotoxic effects (Kreutzberg 1996; Biber et al. 2007; Prinz Mildner 2011; Hellwig et al. 2013). Microglia facilitate the return to homeostasis once an insult is contained by participating in tissue repair, by removing offending agents and cytokines, and by secreting injury healing factors. However, excessive activation of microglia can be cytotoxic through the release of excitatory amino acids and cytokines (Dilger and Johnson 2008). Neurotraumatic (stroke) and neurodegenerative diseases (AD and PD) trigger an immune activation of the brain, which contributes on one hand to brain damage through loss of neurons and on the other hand immune activation may also be involved in tissue repair (Rivest 2009). A common mechanism underlying the central and peripheral effects of exercise may be related to neuroinflammation, which is known to impair growth factor signaling not only in the peripheral tissues, but also in the brain (Cotman et al. 2007). Exercise also induces biochemical alterations in astrocytes (de Senna et al. 2011) and enhances the behavioral performance of animals in spatial memory tasks. Moderate exercise decreases the GFAP content in the rat hippocampus. This is in agreement with a decrease in the number and immunoreactivity of GFAP cells in the rat cerebral cortex after running (Ang et al. 2006). The molecular mechanism associated with decrease in glial fibrillary acidic protein (GFAP) level is not known. However, it is proposed that corticosterone, which is increased due to exercise, can downregulate GFAP expression. In fact, GFAP levels are negatively regulated by glucocorticoids in the rat hippocampus (Nichol et al. 2005). Moreover, it is also speculated that this downregulation of GFAP may be associated with other neuronal changes that accompany GFAP downregulated conditions, such as neurite outgrowth (Menet et al. 2001) supporting the view that decrease in GFAP may be related to indirect signal of neuronal plasticity in the rat hippocampus in response to treadmill exercise .
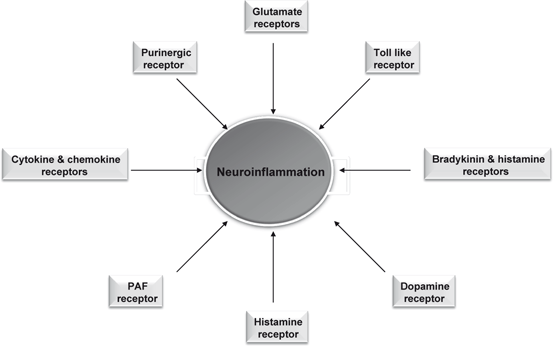
Fig. 5.4
Contribution of various receptors in neuroinflammation
In cardiovascular and cerebrovascular systems exercise mediates it effects by targeting the vasculature (Mora et al. 2007; Green 2009; Padilla et al., 2011). In cardiovascular and cerebrovascular systems, exercise increases endothelium-dependent vasodilation and/or NO bioavailability and improves vascular compliance (Green et al. 2011; Wang et al. 2007). Both chemical and physical factors stimulate endothelium-derived NO production (Vallance and Chan 2001). The formation of NO from L-arginine, molecular oxygen, and NADPH is catalyzed by the phosphorylation of endothelial nitric oxide synthase (eNOS), an enzyme that requires tetrahydrobiopterin, flavin adenine dinucleotide and flavin mononucleotide as cofactors. Shear stress from arterial blood flow, insulin, and acetylcholine initiate pathways to phosphorylate eNOS. Both insulin and shear stress act through a calcium-independent signaling pathway comprising phosphatidylinositol-3-kinase (PtdIns 3K); whereas ACh works through a calcium-dependent pathway. In addition, AMP-activated protein kinase (AMPK), a fuel and stress sensing heterotrimeric serine/threonine kinase, is activated in cultured endothelial cells in response to exercise, shear stress (Fleming et al. 2005), and oxidative stress (Thomas et al. 2002). It is important to note that animal models of insulin resistance show decrease in AMPK activity in skeletal muscle (Fuji et al. 2008; Yu et al. 2004). Changes in AMPK activity may accelerate the progression of insulin resistance and metabolic abnormalities associated with metabolic syndrome (Richter and Ruderman 2009). Exercise can activate skeletal muscle AMPK, effectively resisting diet- or lipid-induced insulin resistance (Hawley and Lessard 2008; Wojtaszewski and Richter 2006) . Once activated, it phosphorylates key metabolic enzymes associated with processes that modulate ATP production and utilization, such as fatty acid oxidation, triglyceride, and cholesterol synthesis (Kemp et al. 2003; Hardie 2011) (Fig. 5.5). Based on these results, it is suggested that AMPK may be an ideal target for the treatment of endothelial cell dysfunction associated with metabolic as well as neurological disorders (Ruderman and Prentki 2004; Manwani and McCullough 2013). Collective evidence suggests that AMPK stands at the crossroads of various metabolic processes and is involved in the metabolic alterations that occur with aging, cell death, and cell survival. For example, activation of AMPK protects endothelial cells, and smooth muscle cells (Xu and Si 2010) under certain stress conditions. However, AMPK activation enhances neuronal death in several models of acute and chronic neurodegeneration, including stroke and Huntington disease (Ju et al. 2011) .
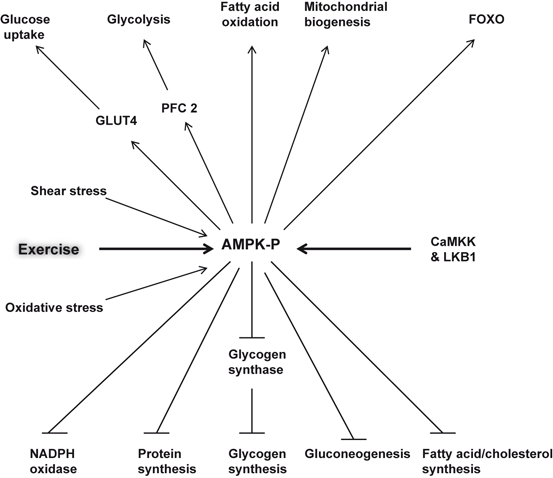
Fig. 5.5
Roles of AMP-activated protein kinase (AMPK) in the brain. Glucose transporter type 4 (GLUT4); Forkhead box O3 (FOXO); liver kinase B1 (LKB1); and 6-phosphofructo-2-kinase (PFK-2)
Exercise influences brain vasculature by increasing the proliferation of brain endothelial cells and angiogenesis throughout the brain (Cotman et al. 2007). The growth factors, such as IGF and VEGF play an important role in the angiogenic and neurogenic effects of exercise on the brain. Thus, running not only upregulates hippocampal IGF gene expression (Carro et al. 2001), but also increases serum levels of both IGF (Carro et al. 2002) and VEGF (Fabel et al. 2003). These growth factors also play an important role in hippocampal neurogenesis . Hippocampal gene transfer of VEGF (Cao et al. 2004) and peripheral infusion of IGF-1 increases neurogenesis (Aberg et al. 2000). Blockade of peripheral VEGF and IGF-1 prevents the increase in running-mediated neurogenesis (Aberg et al. 2000; Trejo et al. 2001) .
Exercise also protects the vasculature through a number of other mechanisms (Katzmarzyk and Lear 2012; Gleeson et al. 2011). Thus, short-term exercise reduces the levels of TNF-α, IL-6, plasminogen activator inhibitor-1 (PAI-1) (Izadpanah et al. 2012), and cell adhesion molecules (Saetre et al. 2011), and protecting against media-intimal hyperplasia (Pahkala et al. 2011) and smooth muscle cell hypertrophy (Pahkala et al. 2011), and strengthening the endothelial barrier (Ding et al. 2006c). TNF-α contributes to the induction and development of insulin resistance through the activation of c-jun N-terminal kinase (JNK), and inhibition of the κ kinase (IKK) and the protein kinase R (PKR) (Gregor and Hotamisligil 2011). Insulin resistance is closely associated with the pathophysiology of type II diabetes and metabolic syndrome, which are risk factor for stroke , Alzheimer disease , and depression (Farooqui et al. 2012; Farooqui 2013). Collective evidence suggests that low-volume/high-intensity resistance exercise can be an important intervention strategy for both the prevention and treatment of the inflammatory state involved in the pathogenesis of metabolic syndrome and related neurological disorders (Farooqui 2013). The high-volume/moderate-intensity swimming exercises promote higher IL-10 expression levels in rats fed the chow diet and the high-fat diet, while resistance training is not able to induce the same adaptations. IL-10 is an important immunoregulatory cytokine with multiple biological effects. In the cytoplasm, IL-10 blocks NF-kB activity at two levels: (a) by suppressing the IKK activity and (b) by preventing binding of NF-kB with DNA (Schottelius et al. 1999). Accumulating evidence suggests that increase in IL-10 expression may be an important protective factor against insulin resistance , obesity related diseases, and metabolic syndrome-related neurological disorders (Waters et al. 2007; Farooqui 2013) . The anti-inflammatory role of exercise (Walsh et al. 2011) is complicated; however, as intense unaccustomed exercise may be associated with increase in cortisol (van Bruggen et al. 2011), C-reactive protein and modest increases in other proinflammatory cytokines (Scott et al. 2011). However, beneficial effects of exercise can be explained through the involvement of proatherogenic adipokines, insulin-sensitizing pathways, or the hemostatic and antioxidant functions of the coronary and cerebral endothelium (Kasapis and Thompson 2005; Hambrecht et al. 2000). Regular exercise attenuates the age-associated increase in oxidative stress and NF-κB activation in animals (Radak et al. 2004), and reduces toll-like receptor 4 (TLR4) signaling which may explain the chronic anti-inflammatory effect of exercise (McFarlin et al. 2005) .
5.4 Exercise-Mediated Neuroprotection in Neurotraumatic, Neurodegenerative, and Neuropsychiatric Diseases
There is growing evidence implicating that exercise can generally slow down aging, prevent chronic and acute neurological diseases, and promote health (Mattson 2000; Haskell et al. 2007). At present no drugs are available for the treatment of neurodegenerative diseases. However, it is becoming increasingly evident that exercise not only reduces the risk of neurological impairment, but also produces beneficial effects in animal models of stroke , AD, PD, dementia and other depression (Eldar and Marincek 2000; Cotman and Berchtold 2002; Smith and Zigmond 2003). The molecular mechanisms associated with the beneficial effects of exercise on neurotraumatic, neurodegenerative, and neuropsychiatric disorders are not fully understood. However, it is well known that neurons are extremely active cells that require a constant supply of energy in order to carry out highly specialized functions, such as regulating the activities of neuronal transmission, receptors, ion channels, transporters and synapses. Mitochondria are vital for producing energy, maintaining homeostasis, and integrity of neuronal function (Farooqui 2010). One of the most important function of the mitochondria is conversion energy from macronutrients (fatty acids, carbohydrates and amino acids) to ATP, through the oxidation of the macronutrients, reducing oxygen to water, and phosphorylating adenosine diphosphate (ADP) to ATP (Schiff et al. 2011; Boveris and Navarro 2008). Defects in mitochondrial function, especially in respiration result in debilitating, possibly fatal abnormalities. For example, mitochondrial dysfunction has been linked to neurotraumatic, neurodegenerative and neuropsychiatric diseases (Farooqui 2010; Clay et al. 2010). These disorders are accompanied by mitochondrial dysfunction, impairment in the respiratory complexes, increase in oxidative stress , induction of neuroinflammation, and neuronal loss in specific area of the brain (Farooqui 2010). Detailed studies have indicated that a reduction in complex IV activity occurs in AD, which is characterized by neuronal death in the nucleus basalis and hippocampus (Chagnon et al. 1995). AD is also characterized by increase in production of proinflammatory lipid mediators (prostaglandins and platelet activating factor) and increased expression of cytokines (Farooqui 2010). A decrease in complex I activity has been reported to occur in the substantia nigra of postmortem samples in PD, which is characterized by neurodegeneration in substantia nigra (Mizuno et al. 1989; Farooqui 2010). Increase in lipid mediators and platelet activation factor also occurs in PD. In stroke , neuronal damage occurs in a relatively homogenous population of neurons in a specific area. For example, an infarct might involve the thalamus, hippocampus, and striate visual cortex, affecting three or more very different neuronal populations including neurons, oligodendrocytes, astrocytes, and endothelial cells (Farooqui 2010). In ischemic injury, generation of ROS blocks mitochondrial respiratory chain. This process stimulates formation of transition pore leading to the release apoptosis-related protein following ischemic brain injury (Moroa et al. 2005). In animal models of stroke , AD, PD, dementia, and depression, exercise lowers the level of ROS, prevents the decline in cytochrome oxidase activity, improves mitochondrial functions, and elevates neurological performance as well as neuronal cell survival (Conley et al. 2007; Navarro and Boveris 2007). In addition, moderate exercise increases the release of β-endorphin in brain and promotes the biogenesis of mitochondria in distant organs.
5.4.1 Beneficial Effects of Exercise in Seniors
Brain aging is accompanied by a decrease in functional capacity and increase in vulnerability of neurons due to inability to organize neural cell function . Brain uses oxygen and glucose to drive cellular energy and metabolism necessary for the maintenance and restoration of ionic gradients, but these processes are diminished with age (Anderson et al. 2010). Decrease in the maintenance of ionic gradients results in neuronal cell death and brain damage. Cell death is particularly notable in neurons because they store information, which is required over a lifetime (Anderson et al. 2010). Brain function depends upon not only on the ability of the brain to maintain neural cell organization through metabolism and cerebral blood flow, but also on the availability of energy.
Exercise increases metabolism and cerebral blood flow leading to increase in synaptic plasticity, synaptic strength, neurogenesis , and angiogenesis (Cotman et al. 2007). Molecular mechanisms associated with these processes are not clear. However, as stated above that exercise reduces the risk of cognitive decline by (a) increasing blood flow to the brain; (b) inhibiting neuroinflammation; (c) increasing production of trophic factors; and (d) increasing neurogenesis. Collective evidence suggests that exercise may produce widespread effects such as the up-regulation of vasculature and neurotrophins both centrally and peripherally, leading to improved delivery of oxygen and other nutrients to brain tissues etc., and generalized cognitive benefits (Farooqui 2013). It should be noted here that most of the findings on the effect of exercise on brain have emerged from basic research on animal models of neurodegenerative diseases, and little is known about what happens in the human brain (van Praag et al. 1999; Neeper et al. 1996). Recent studies have indicated that older adults who participated in an aerobic exercise intervention for 6 months boosted brain volume, in both gray and white matter, suggesting that cognitive function in human brain can be enhanced through aerobic activity (Colcombe et al. 2006). Based on this information, it can be proposed that exercise is one of the most effective interventions for aging and neurodegenerative diseases. It can simultaneously slow age-related decline in neuronal metabolic capacity and maintain cerebrovascular support for neuronal metabolism (Jedrziewski et al. 2007; Laurin et al. 2001) .
5.4.2 Beneficial Effects of Exercise in Stroke Patients
Stroke (ischemic injury) is a highly dynamic multifactorial metabolic insult caused by severe reduction or blockade in cerebral blood flow due to the formation of a thrombus (clot) or embolism . This blockade not only decreases oxygen and glucose delivery to the brain tissue, but also results in the breakdown of blood brain barrier (BBB), build-up of potentially toxic products in the brain, and mitochondrial dysfunction (Farooqui 2010). Subsequently, stroke-mediated neural cell injury cascade not only involves excitotoxicity, oxidative stress , apoptosis and inflammatory processes, but also activation of glial cells and infiltration of leukocytes. These processes trigger the pathological signal transduction pathways that ultimately cause irreversible neuronal injury in the ischemic core within minutes of the onset (Dirnagl et al. 1999). Age is an important risk factor for stroke, yet aging is rarely considered in preclinical models of ischemic stroke. Pathophysiological mechanisms in the brain response to an ischemic insult in old age are not fully understood. Most preclinical stroke studies have been performed in young animal models, which do not replicate the pathophysiology of the aged brain (Di Napoli and Shah 2011). This has been one of the criticisms of preclinical stroke neuroprotection studies (Ford 2008). At the molecular level, stroke is accompanied by the influx of Ca2+ , release of glutamate, increase in eicosanoids and platelet activating factor, and activation of phospholipases, cyclooxygenases, lipoxygenases, calpains, matrix metalloproteinases (MMPs), and caspases along with increase in expression and secretion of cytokines and chemokines (Farooqui 2010) . Cytokines up-regulate cellular adhesion molecules (CAMs). CAMs are responsible for the adhesion and migration of the leukocytes. Leukocytes roll on the endothelial surface and then adhere to the endothelial cells. The interactions between leukocytes and the vascular endothelial cells are mediated by three main groups of CAMs: the selectins, the immunoglobulin gene superfamily, and the integrins. Selectins, especially E- and P-selectins are upregulated and mediate leukocyte rolling and recruitment during the early stages of ischemic injury (Zhang et al. 1998). Phospholipases A2 and cyclooxygenases I and II, and lipoxygenases are involved in breakdown of neural membrane phospholipid. The spatial and temporal upregulation of enzymic activities (phospholipases, cyclooxygenases, lipoxygenases, calpains, and caspases) and cytokines and their receptors depends on the ischemic model used (Lai and Todd 2006). MMPs are involved in the breakdown of the microvascular basal lamina, which results in the disruption of the BBB (Heo et al. 1999). These changes are most prominent in the core infarct, where neuronal damage is maximal. Other enzymes along with MMP-9 play an important role in the progression of the cerebral infarct. As mentioned earlier chapters, main cytokines involved in neuroinflammation are interleukins (IL), IL-1, IL-6, IL-10, and TNF-α. Cytokines are not only responsible for the stimulation of phospholipases A2 and cyclooxygenases, but also for the initiation and regulation of the inflammatory response and play an important role in leukocyte infiltration into the ischemic regions of the brain (Kaushal and Schlichter 2008) .
Studies on in vivo and in vitro models of stroke have indicated that brain uses several proteins and signaling pathways to protect neurons against ischemic injury (Farooqui 2010). These include neurotrophic factors (BDNF, GDNF and VEGF), Nuclear factor erythroid-related factor 2 (Nrf2), protein chaperones (heat shock protein 70, Hsp70) and glucose regulated protein 78 (GRP78); antioxidant enzymes (heme oxygenase-1), and the regulator of mitochondrial biogenesis PGC-1α. It is shown that increased expression and production of above mentioned factors not only protects neurons from stroke-mediated neuronal injury, but also increases the rate of neurogenesis in rodents (Lee et al. 2002; Kernie and Parent 2010). As stated earlier, Nrf2 is a transcription factor that regulates a group of antioxidant genes that act to remove ROS through sequential enzymatic reactions (Nguyen et al. 2009). Studies in animal models of stroke have indicated that increasing Nrf2 activity is highly neuroprotective stroke-mediated neuronal damage (Johnson et al. 2008) Thus, administration of tert-butylhydroquinone (tBHQ), a well known Nrf2 inducer significantly improves sensorimotor and histological outcome in two models of I/R in rats and mice (Shih et al. 2005). It is also reported that Nrf2 activation before stroke salvages the cortical penumbra but not the stroke core following ischemic injury.
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Neurochemical Aspects of Oxidative and Nitrosative Stress
 Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
 Effect of Exercise on Oxidative Stress in Neurological Disorders
Effect of Exercise on Oxidative Stress in Neurological Disorders
 Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
Contribution of Transcription Factors and Genes in the Induction of Oxidative Stress
 Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
 Contribution of Dietary Fat in Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


